 8 (926) 907 94 08
8 (926) 907 94 08
Всё на свете является чудом!
Множественное выравнивание
a. Получение 5–10 гомологов своего белка, используя BLAST на NCBI.
- для поиска гомологов белка THIS_BACSU мы запустили BLAST, ограничив выдачу таксоном Bacteria и установив порог на E-value, равный 0,001. Но на выходе мы не то что не получили слишком родственных белков, у нас вообще ничего не нашлось. Поэтому продолжили поиск уже другим способом.
- если в первом случае мы искали по SwissProt, то уже после отсутствия находок стали искать по nr и конечно же нашли гомологи. Из огромного количества гомологов выбрали 5 и записали их в файл с адресами последовательностей.
- затем командой (seqret @myproteins.list myproteins.fasta) получили эти последовательности в fasta-формате.
b. Построение множественного выравнивания моего белка и всех найденных гомологов.
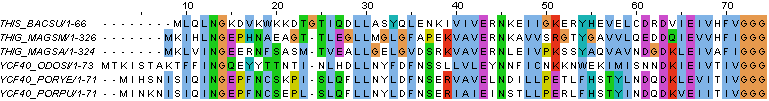
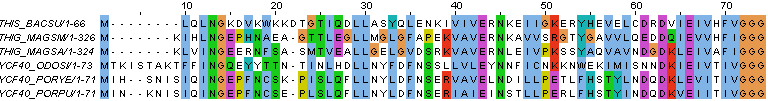
- открыв выравнивание в JalView, через меню File загрузили файл с последовательностями в fasta-формате
- затем через меню "Web Service - Alignment" построили выравнивания с помощью разных программ (Clustal и TCoffee)
- построив выравнивания, раскрасили их с помощью ClustalX - одной из схем раскраски аминокислотных остатков.
↓
→ выравнивание с помощью Clustal
↓
→ выравнивание с помощью программы TCoffee
c. Описание структуры выравнивания.
Для того, чтобы лучше разобраться в выравнивании и "прочувствовать" участки с разной долей консервативных позиций, было сделано несколько раскрасок выравнивания в зависимости от порога консервативности (100, 40 и 20):
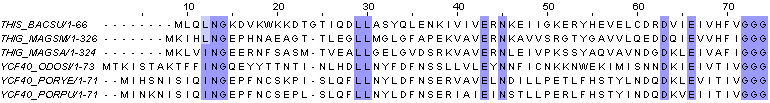 ->
порог=100
->
порог=100 ->
порог=40
->
порог=40 ->
порог=20
->
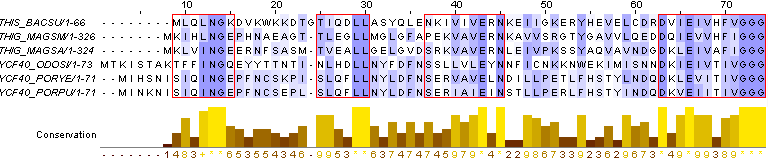
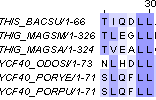
порог=20Теперь для последнего случая выделим блоки с повышенной долей консервативных позиций, в этом нам помогла разметка Conservation, а также интенсивность окраски в самом выравнивании:
|
Координаты по столбцам выравнивания |
Координаты по остаткам моего белка |
Скрин-шоты |
|
9 - 15 |
1 - 7 |
|
|
25 - 30 |
17 - 22 |
|
|
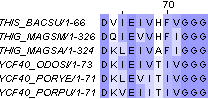
37 - 45 |
29 - 37 |
|
|
63 - 74 |
55 - 66 |
|


{kind=link}
{kind=link}
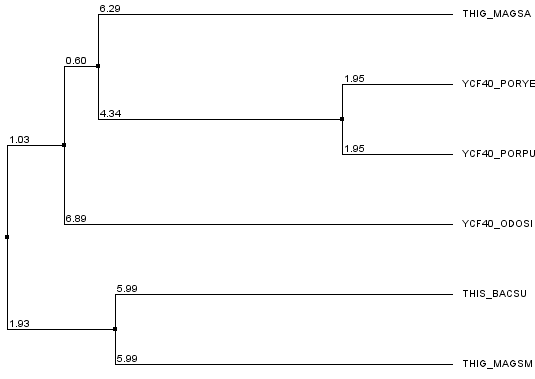
К описанию выравнивания добавим филогенетическое дерево (процент идентичности, среднее расстояние), где данные не противоречат таксономическому положению организмов:
Также можно утверждать, что на участках 1-7 и 75-334 выравнивание не имеет биологического смысла. Во втором случае выделяем такой большой участок из-за того что среди выбранных нами белков - 4 имеют длину последовательности до 75 аминокислотных остатков, а у 2-х белков длина составляет уже 335 а.о. Как раз на картинке мы видим как, начиная с 75-ого столбца, выравниваются только два длинных белка.
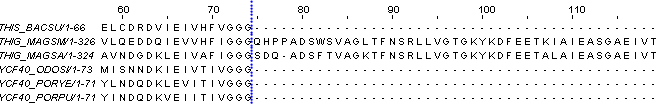 - - -
- - -
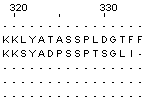
Так что для этих двух белков выравнивание безусловно несёт в себе биологический смысл, но во множественном выравнивании с другими белками мы этот продолжительный участок не рассматриваем.
d. Указание групп сходных аминокислот, которые образуют в моем выравнивании функционально консервативные позиции.
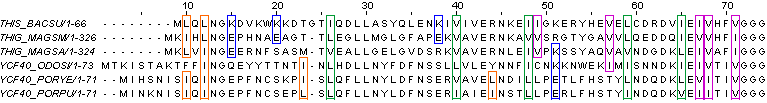
Функционально консервативные позиции в выравнивании образуют следующие группы сходных аминокислот (в скобках указано число таких групп в выравнивании):
LIV - [5]
LI - [8]
IV - [4]
KE - [4]
Можно, конечно, привести ещё примеры таких групп, но мы выбрали самые явные и часто встречающиеся.
2. Программа Muscle.
Чтобы получить последовательности малых дельта-антигенов из банка Swiss-Prot, мы воспользовались SRS. Все дельта-антигены происходят из вирусов рода "Deltavirus" и имеют в описании слово "delta"; малые дельта-антигены в описании имеют ещё слово "small". Поэтому в SRS создаём запрос к банку Swiss-Prot, написав:
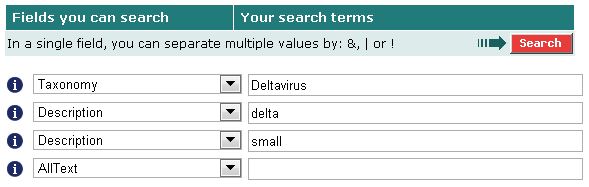
17 найденных последовательностей сохранили в fasta-формате.
Далее открываем этот файл в JalView. После чего с помощью программы muscle выравниваем невыровненные последовательности, выполняя команду (muscle -in delta.fasta -out delta_aligned.fasta). На выходе получаем файл с выровненными последовательностями в fasta-формате.
То же выравнивание можно было получить и с помощью muscle в JalView. Отличие лишь в том, что последовательности остаются в заданном порядке (при параметрах по умолчанию), а в первом случае - они выдавались по увеличению длины последовательностей.
Используя JalView, раскрашиваем выравнивание по BLOSSUM62 с порогом 30.
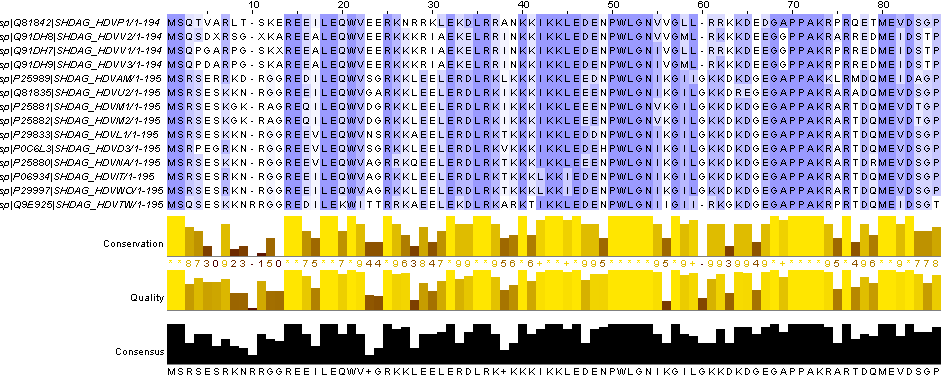
Как мы видим консервативных участков достаточно много.
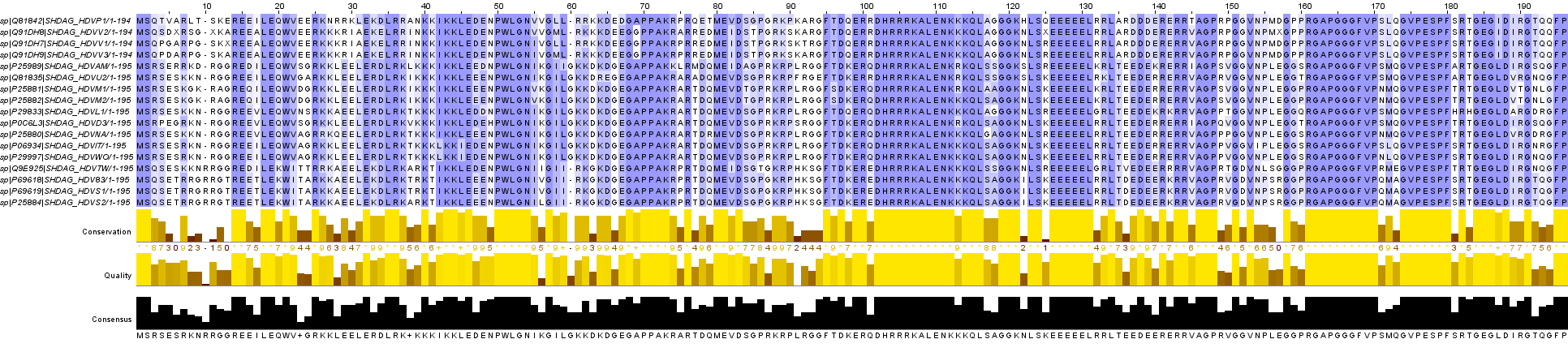{kind=link}
Имеются идентичные с большой протяженностью: 50-54, 70-73, 102-112, 126-131, 161-170, 174-180 (координаты для выравнивания).
А в качестве функционально консервативных можно выделить: KK, RK, GG, LS и т.д.
Выравнивание сохранили в формате msf, а также в формате jar (так как он позволяет сохранять выравнивания с дополнительной информацией, например окраской)
| Главная | Об авторе | Учебные семестры | Проекты автора | Друзья | Ссылки партнеров | Extra | Контакты |
Mneff © 2011-2012