 8 (926) 907 94 08
8 (926) 907 94 08
Всё на свете является чудом!
Реконструкция филогенетических деревьев
Используя таксономический сервис NCBI, определили к каким таксонам относятся отобранные ранее бактерии.
Полученные данные представлены в виде таблицы:
| Название | Мнемоника | Таксономия | ||||||
| Домен | Тип | Класс | Порядок | Семейство | Род | Группа | ||
| Bacillus anthracis | BACAN | Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae | Bacillus | Bacillus cereus group |
| Bacillus subtilis | BACSU | Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae | Bacillus | Bacillus subtilis group |
| Clostridium botulinum | CLOB1 | Bacteria | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | - |
| Clostridium tetani | CLOTE | Bacteria | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | - |
| Staphylococcus aureus | STAA1 | Bacteria | Firmicutes | Bacilli | Bacillales | Staphylococcaceae | Staphylococcus | - |
| Staphylococcus epidermidis | STAES | Bacteria | Firmicutes | Bacilli | Bacillales | Staphylococcaceae | Staphylococcus | - |
| Streptococcus pyogenes | STRP1 | Bacteria | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | - |
| Streptococcus pneumoniae | STRPN | Bacteria | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | - |
Также на NCBI имеется инструмент, строящий таксономические деревья (для этого достаточно лишь вписывать интересующие нас бактерии или другие организмы).
Ниже представлено таксономическое дерево для отобранных нами бактерий:

Чтобы наглядно увидеть соответствие таксономического и филогенетического деревьев, ниже приведено ранее полученное филогенетическое дерево:
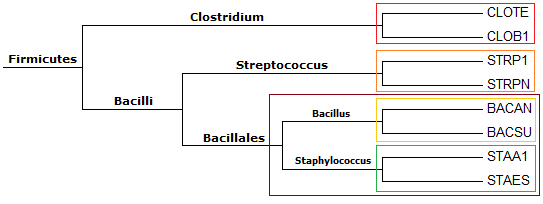
Нетривиальные ветви, выделяющие таксоны, обведены цветными рамками как в таксономическом, так и в филогенетическом деревьях.
Как видно, все 5 нетривиальных ветвей в филогенетическом дереве выделяют таксоны.
Ниже представлены нетривиальные ветви и таксоны, которые они выделяют:
1. {CLOTE, CLOB1} против {STRP1, STRPN, BACAN, BACSU, STAA1, STAES} → род Clostridium (класс Clostridia) и класс Bacilli
2. {STRP1, STRPN} против {CLOTE, CLOB1, BACAN, BACSU, STAA1, STAES} → род Streptococcus (порядок Lactobacillales)
3. {BACAN, BACSU} против {CLOTE, CLOB1, STRP1, STRPN, STAA1, STAES} → род Bacillus (семейство Bacillaceae)
4. {STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN, BACAN, BACSU} → род Staphylococcus (семейство Staphylococcaceae)
5. {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN} → порядок Bacillales
2. Реконструкция филогенетического дерева (по семейству белков с определённой функцией)
Из банка Swiss-Prot получаем последовательности белков с функцией "Фактор элонгации трансляции G" (мнемоника - EFG) при помощи команды seqret:
seqret sw:efg_bacan
Затем объединяем полученные файлы с последовательностями белков в один файл с помощью команды cat:
cat bacan.fasta bacsu.fasta clob1.fasta clote.fasta staa1.fasta staes.fasta strp1.fasta strpn.fasta > all_bacteria.fasta
Далее редактируем названия последовательностей, оставляя только мнемонику видов, после чего выравниваем эти последовательности с помощью программы muscle:
muscle -in all_bacteria.fasta -out alignment_all_bacteria.fasta
Полученное выравнивание открываем через программу JalView и представляем его в «блочной» форме с блоками шириной 100 остатков и раскраской по проценту идентичности:
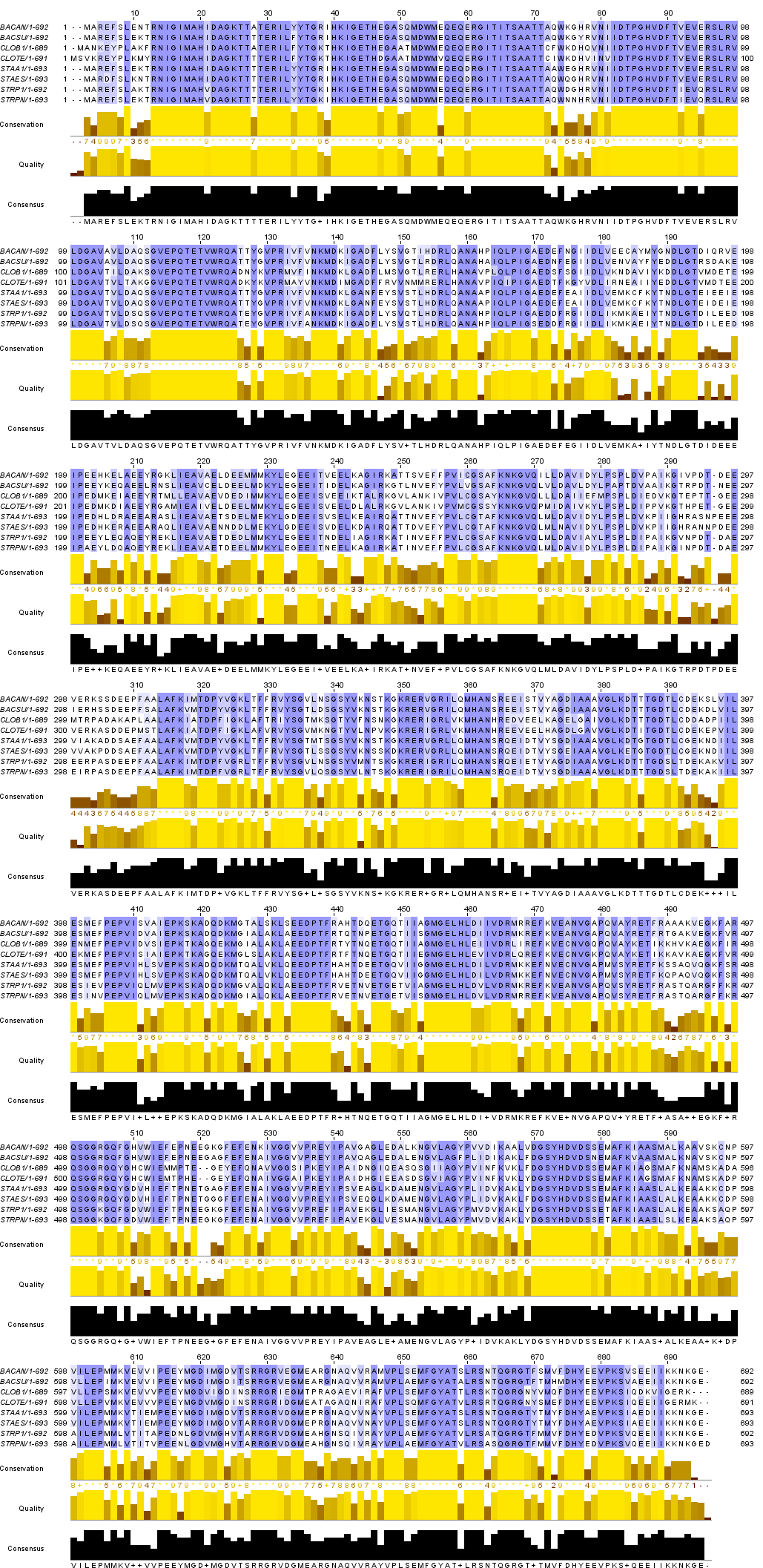
Проект Jalview сохранили в формате jar.
3*. Поиск диагностических позиций выравнивания
В выравнивании можно найти так называемые диагностические позиции, которые показывают относится ли та или иная последовательность выравнивания к некоторому таксону или нет.
По этим позициям мы определим и к какому конкретно таксону относится эта последовательность.
Для этого просматриваем каждый столбец выравнивания и отмечаем те столбцы, где встречаются одинаковые буквы, но именно для конкретных бактерий, которые образуют таксон.
Например, в столбце 106 у первых шести последовательностей тимин (T), а у 7 и 8 последовательности аденин (А). В свою очередь, две последние последовательности - это BACAN и BACSU, которые образуют таксон. Таким образом, можно диагностировать и остальные участки выравнивания.
Ниже приведено выравнивание, где продиагностированно 4 типа позиций, которые дают понять о соответственно 4-х таксонах (одна из диагностических позиций определяет даже 2 таксона):

На самом деле, в этом выравнивании диагностических позиций больше, чем отмечено на картинке. Но это сделано не просто так.
Для того, чтобы наглядно разобраться и лучше понять принцип, были выделены только "чистые" позиции, то есть те, в которых выделяются буквы, характерные для какого-то определённого таксона, при этом остальные буквы в этой позиции тоже одинаковые, но не совпадают с буквами, определяющими таксон. Зато "чистые" позиции выделены все, поэтому можно уверенно сказать, что позиций, выделенных жёлтыми стрелочками, больше всего, зелёных и красных примерно поровну, а голубых практически нет.
Исходя из этого, можно сделать вывод, что род Clostridium и класс Bacilli (позиции под жёлтыми стрелочками) диагностируются лучше по сравнению с другими. Есть предположение почему это так - если посмотреть на филогенетическое дерево (повторно показано ниже), то видно, что таксоны Clostridium и Bacilli стоят выше по уровню, чем остальные, поэтому можно предположить, что и диагностируются они в выравнивании заметно чаще.
4. Реконструкция филогенетического дерева (методами, доступными из JalView)
Необходимо реконструировать филогенетическое дерево полученных ранее организмов, методами из программы JalView.
Эта программа располагает четырьмя методами построения деревьев. Все они доступны из меню (Calculate > Calculate Tree).
Изображения импортированы программой Mega, после открытия соответствующих файлов.
1) Average Distance Using % Identity
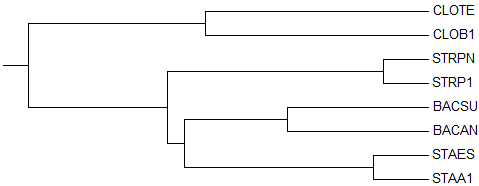 →
1-ое реконструированное дерево
→
1-ое реконструированное дерево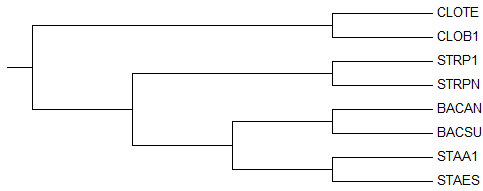 → правильное
дерево
→ правильное
деревоКак видно, оба дерева идентичны (отличия лишь в порядке названий).
2) Neighbour Joining Using % Identity
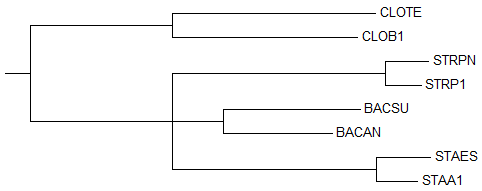 →
2-ое реконструированное дерево → правильное дерево
→
2-ое реконструированное дерево → правильное деревоВ реконструированном дереве ветви {STRPN, STRP1}, {BACSU, BACAN}, {STAES, STAA1} отходят от одной ветки сразу, а в правильном дереве ветки {BACSU, BACAN} и {STAES, STAA1} соединены сначала между собой.
Из-за допущенной ошибки в реконструированном дереве отсутствует нетривиальная ветвь {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN}, которая безусловно присутствует в правильном дереве.
3) Average Distance Using BLOSUM62
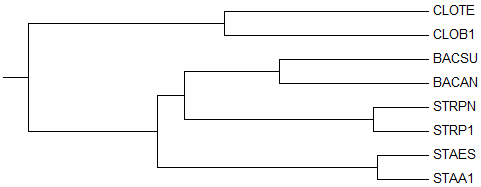 →
3-е реконструированное дерево →
правильное дерево
→
3-е реконструированное дерево →
правильное дерево В реконструированном дереве ветки {STRPN, STRP1} и {STAES, STAA1} перепутаны местами, в отличие от правильного дерева. Если ветку {STAES, STAA1} поместить под общую ветку с {BACSU, BACAN}, а {STRPN, STRP1}, наоборот, выделить в отдельную, то мы получим правильное дерево.
В реконструированном дереве выделяется нетривиальная ветвь {BACSU, BACAN, STRPN, STRP1} против {CLOTE, CLOB1, STAES, STAA1}, которая, в свою очередь, отсутствует в правильном дереве.
Вместо этой неправильной нетривиальной ветви в правильном дереве присутствует нетривиальная ветвь {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN}.
4) Neighbour Joining Using BLOSUM62
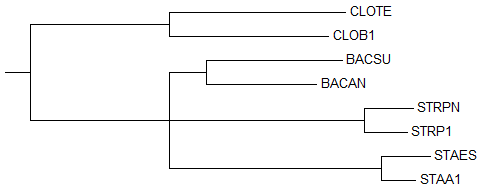 →
4-ое реконструированное дерево →
правильное дерево
→
4-ое реконструированное дерево →
правильное деревоВ этом реконструированном дереве та же ошибка, что и во втором: ветви {STRPN, STRP1}, {BACSU, BACAN}, {STAES, STAA1} отходят от одной ветки сразу, а в правильном дереве ветки {BACSU, BACAN} и {STAES, STAA1} соединены сначала между собой.
Как и во втором реконструированном дереве, из-за допущенной ошибки отсутствует нетривиальная ветвь {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN}, которая безусловно присутствует в правильном дереве.
5. Реконструкция филогенетического дерева (методом "Maximum Parsimony")
Ещё одно реконструированное дерево можно получить, импортируя выровненные последовательности в программу MEGA и использовав метод "Maximum Parsimony" (в переводе с англ. "Максимальная Экономия").
Ниже представлено реконструированное дерево, построенное с помощью этого метода и укорененное в ветвь {CLOB1, CLOTE} против {STAA1, STAES, STRP1, STRPN, BACAN, BACSU}:
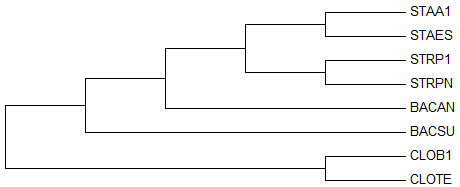 → 5-ое
реконструированное дерево → правильное дерево
→ 5-ое
реконструированное дерево → правильное деревоПро то как исправить реконструированное дерево говорить не имеет смысла, так как уж больно много допущено ошибок.
Три правильные нетривиальные ветви у реконструированного дерева присутствуют:
- {CLOTE, CLOB1} против {STRP1, STRPN, BACAN, BACSU, STAA1, STAES}
- {STRP1, STRPN} против {CLOTE, CLOB1, BACAN, BACSU, STAA1, STAES}
- {STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN, BACAN, BACSU}
Ещё две нетривиальные ветви есть у правильного дерева, но отсутствуют у реконструированного:
- {BACAN, BACSU} против {CLOTE, CLOB1, STRP1, STRPN, STAA1, STAES}
- {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN}
Также у реконструированного дерева ветви {BACAN} и {BACSU} не объединены в нетривиальную ветвь, а, напротив, выделены в отдельные тривиальные ветви.
6*. Сервис "TreeTop"
Используя сервис TreeTop (созданный в НИИФХБ имени А.Н. Белозерского) можно реконструировать филогенетическое дерево по выравниванию в fasta-формате.
Ниже приведено дерево, построенное по выравниванию, сделанному ранее:
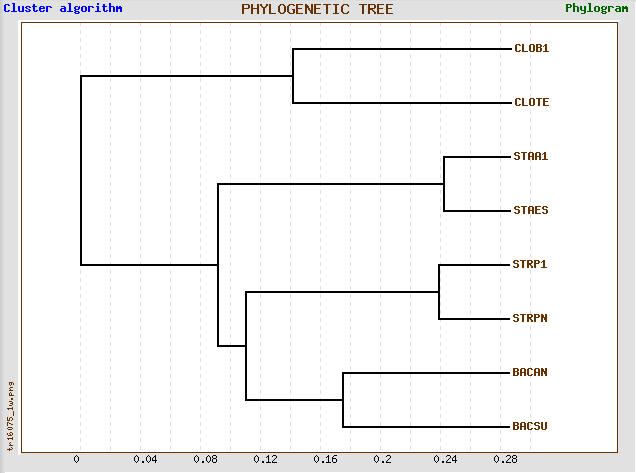 → 6-ое
реконструированное дерево
→ правильное дерево
→ 6-ое
реконструированное дерево
→ правильное деревоВ реконструированном дереве вновь допущена ошибка, на этот раз такая же, как и в 3-м дереве (из JalView): ветки {STRPN, STRP1} и {STAES, STAA1} перепутаны местами, в отличие от правильного дерева. Если ветку {STAES, STAA1} поместить под общую ветку с {BACSU, BACAN}, а {STRPN, STRP1}, наоборот, выделить в отдельную, то мы получим правильное дерево.
В реконструированном дереве выделяется нетривиальная ветвь {BACSU, BACAN, STRPN, STRP1} против {CLOTE, CLOB1, STAES, STAA1}, которая, в свою очередь, отсутствует в правильном дереве.
Вместо этой неправильной нетривиальной ветви в правильном дереве присутствует нетривиальная ветвь {BACAN, BACSU, STAA1, STAES} против {CLOTE, CLOB1, STRP1, STRPN}.
Впечатления о работе сервиса TreeTop:
Есть полезная опция отправки результатов на адрес электронной почты. Можно заранее выбрать формат дерева и картинок. На странице с результатами содержится информация о параметрах алгоритма, выравнивание, реконструированные деревья в текстовом и графическом форматах, матрица расстояний и даже скобочные формулы реконструированных деревьев.
Всё это конечно идёт в плюс, поэтому за столько краткое знакомство с сервисом, особенных минусов найдено не было. Единственное, привычнее получать графическое филогенетическое дерево в более компактном виде, как, например, используя программы JalView или MEGA.
| Главная | Об авторе | Учебные семестры | Проекты автора | Друзья | Ссылки партнеров | Extra | Контакты |
Mneff © 2011-2013