Анализ хроматограмм, полученных методом Сэнгера
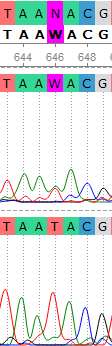
Для "прямой" хроматограммы (39_F.ab1):
-длина нечитаемого участка в начале 57 нуклеотидов (1-57)
-длина нечитаемого участка в конце 24 нуклеотида (693-716)
-отношение интенсивности шума к интенсивности сигнала примерно 0,25
-шум превалирует в конце прочтения, но есть шумные участки в середине и конце
-исходный файл
Для "обратной" хроматограммы (39_R.ab1):
-длина нечитаемого участка в начале 57 нуклеотидов (1-57)
-длина нечитаемого участка в конце 52 нуклеотида (667-718)
-отношение интенсивности шума к интенсивности сигнала примерно 0,10
-шума больше в начале прочтения и в самом конце, участок 240-560 почти лишён шума
-исходный файл
Для обрезки нечитаемых концов и преобразования обратного прочтения я использовал написанный мной скрипт. При выделении нечитаемых начальной и концевой областей я ориентировался на ранжирование символов четвёртой строки на странице в Википедии, интуитивно задав порог читаемости (при желании его можно легко изменить)
Итоговое выравнивание в формате .fasta; первая последовательность - консенсус, полученный до редактирования хроматограмм, вторая и третья - выровненные по нему и исправленные по хроматограммам последовательности
К сожалению, у меня не получилось сделать замены маленькими буквами, UGene не даёт
:(

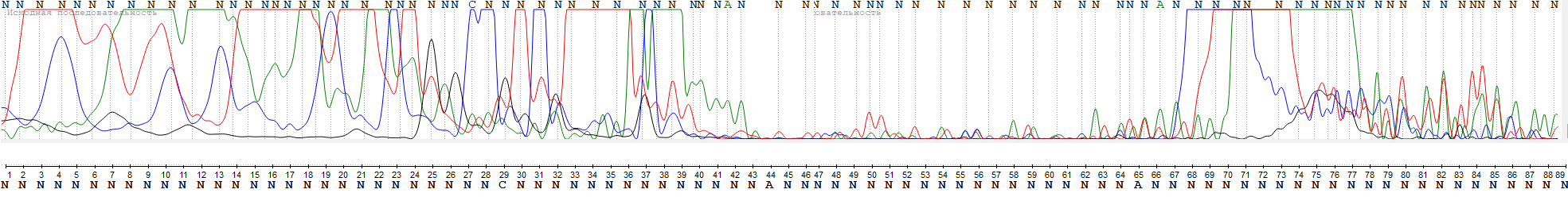
Здесь доступно изображение плохой хроматограммы (WS2943_SP6R)
{kind=link}
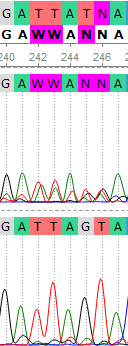
Первые 40 нуклеотидов, как и в большинстве случаев, нечитаемы, что отражает характер посадки праймера на близлежащий участок
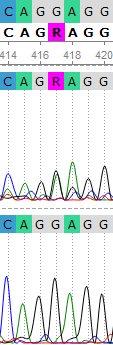
Нуклеотиды 40-66 имеют сравнительно нормальную читаемость (несмотря на это, программа почти везде поставила N, хотя, на мой взгляд, при увеличении масштаба некоторые пики вполне различимы), однако испытывают беды с масштабированием как относительно начальной/конечной областей, так и друг с другом
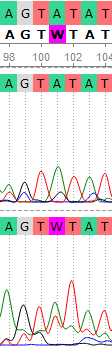
В позициях 67-78 наблюдаются пятна красителей, мешающие обзору
Позиции 79-89 имеют приемлемую читаемость, однако программа снова не определила большинство нуклеотидов