Task 1
Prediction of the secondary structure of observed tRNA
| Structure area | find_pair | einverted | Zuker algorithm |
| Acceptor stem | 5'-2-7-3' 3'-71-66-5' 6 pairs | 6 pairs out of 7 | 6 pairs out of 7 |
| D-loop | 5'-49-53-3' 3'-65-61-5' 5 pairs | 0 pairs out of 4 | 5 pairs out of 4 |
| T-loop | 5'-39-43-3' 3'-31-27-5' 5 pairs | 0 pairs out of 5 | 5 pairs out of 5 |
| Anticodon loop | 5'-10-12-3' 3'-25-23-5' 3 pairs | 0 pairs out of 4 | 3 pairs out of 4 |
| Total amount of canonical pairs | 19 | 6 | 19 |
SEQUENCE: Score 18: 6/6 (100%) matches, 0 gaps
1 ggggta 6
||||||
69 ccccat 64
Gap penalty [12]: 10
Minimum score threshold [50]: 10
Match score [3]:
Mismatch score [-4]:
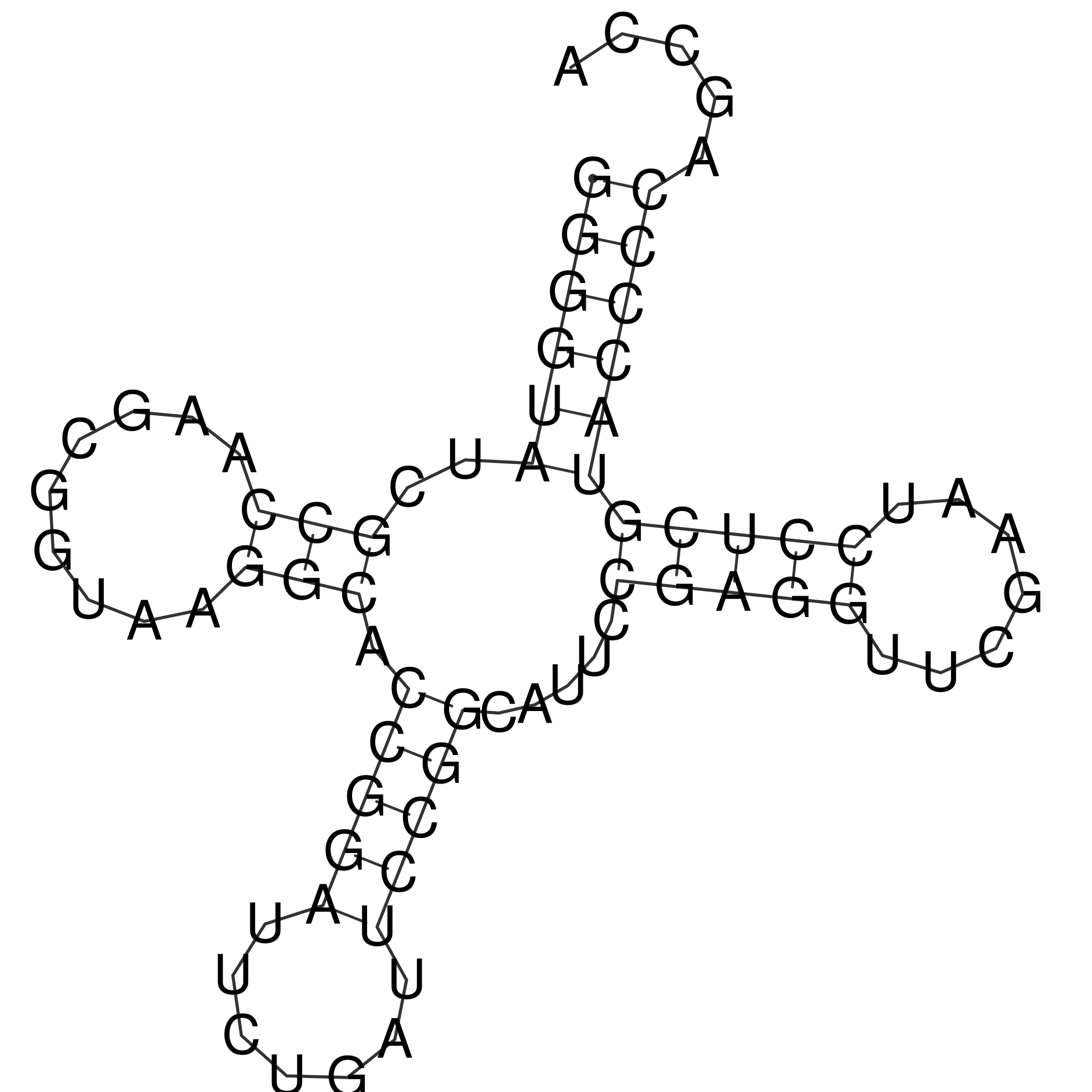
Outcome of einverted program was empty with standard parameters, so custom ones were used. RNAfold outcome was very close to the reality from the very first attempt with default parameters. Figure 1 displays graphical image of suggested structure and probability distribution of nucleotide pair formation. According to Table 1, the Zuker algorithm was the most accurate.
Task 2
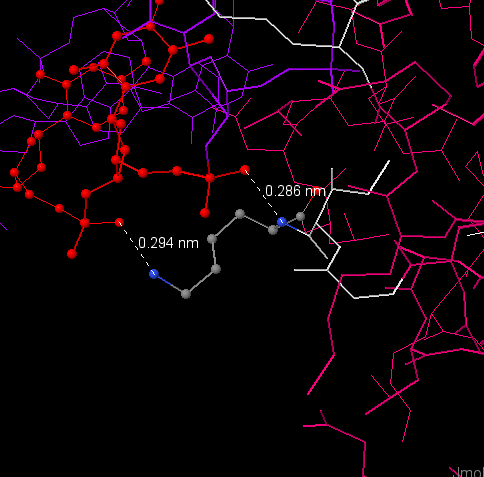
DNA-protein contacts in observed structure
| Protein contacts (chains M, R and Y) | Polar | Non-polar | Total amount |
| with the residues of 2'-deoxyribose | 1 | 20 | 21 |
| with residues of phosphoric acid | 8 | 0 | 8 |
| with nucleotide residues of major groove | 2 | 3 | 5 |
| with nucleotide residues of minor groove | 2 | 0 | 2 |
Task 3
Nucplot
According to nucplot outcome, amino acid residue with the largest number of contacts is LYS 79, having 2 contacts with T(32) and C(31). It is also sensible to assume that LYS 79 is the most important residue for DNA recognition considering the fact it interacts with two nucleotides.
The lack of image from nucplot is explained by incorect work of program: "In some cases, the graph-matching algorithm of HBADD fails and some of the atom-name mappings between the PDB file and the HET Group Dictionary are missed. This flaw will be rectified soon."