Command table
Command table
| Command | Description |
|---|---|
| fastqc chr9_1.fastq | FastQC reads a set of sequence files and produces from each one a quality control report consisting of a number of different modules, each one of which will help to identify a different potential type of problem in your data. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9_1.fastq 9.1cut.fastq TRAILING:20 MINLEN:50 | This command cuts off parts with average quality less than 20 and removes reads that are less than 50 nucleotides long. Quality changes are presented in Fig.1 and 2 |
| hisat2-build chr9.fasta r_chr9_ind | Indexing of reference sequence |
| hisat2 -x r_chr9_ind -U 9.1cut.fastq --no-spliced-alignment --no-softclip -S chr9_alig.sam --summary-file hisat2.txt | Alignment of reads and reference sequence |
| samtools view -t chr9.fasta.fai -b chr9_alig.sam -o chr9_alig.bam | Conversion of alignment into binary format |
| samtools sort chr9_alig.bam -T temp -o chr9_sort.bam | Alignment sorting by the coordinate in the reference |
| samtools index chr9_sort.bam | Indexing of sorted file |
| samtools mpileup -uf chr9.fasta chr9_sort.bam -o polymorph9.bcf | Generating of file containing polymorphisms |
| bcftools call -cv polymorph9.bcf -o polymorph9.vcf | Generating of file containing list of differences between reference and reads |
| convert2annovar.pl -format vcf4 polymorph9.vcf -outfile polym.avinput | Change file format to suitable for annovar |
| annotate_variation.pl -out poly_refgene -build hg19 polym.avinput /nfs/srv/databases/annovar/humandb.old/ | Annotate SNPs in refgene |
| annotate_variation.pl -filter -out poly_dbsnp -build hg19 -dbtype snp138 polym.avinput /nfs/srv/databases/annovar/humandb.old/ | Annotate SNPs in dbsnp |
| annotate_variation.pl -filter -out poly_dbsnp -buildver hg19 -dbtype 1000g2014oct_all polym.avinput /nfs/srv/databases/annovar/humandb.old/ | Annotate SNPs in 1000 genomes |
| annotate_variation.pl -regionanno -out poly_gvas -build hg19 -dbtype gwasCatalog polym.avinput /nfs/srv/databases/annovar/humandb.old/ | Annotate SNPs in Gwas |
| annotate_variation.pl -filter -out poly_clinvar -buildver hg19 -dbtype clinvar_20150629 polym.avinput /nfs/srv/databases/annovar/humandb.old/ | Annotate SNPs in Clinvar |
Task 1. Reads preparation
Amount of reads didn't change dramatically: from 10701 to 10536 (165 were dropped). According to the command was used, reads whose length was less than 50 (after cutting off low quality parts) were dropped.
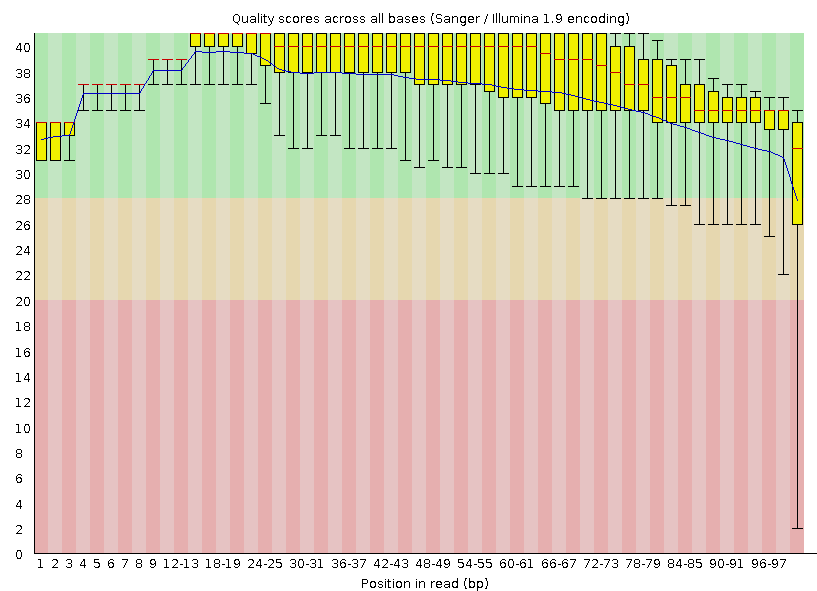
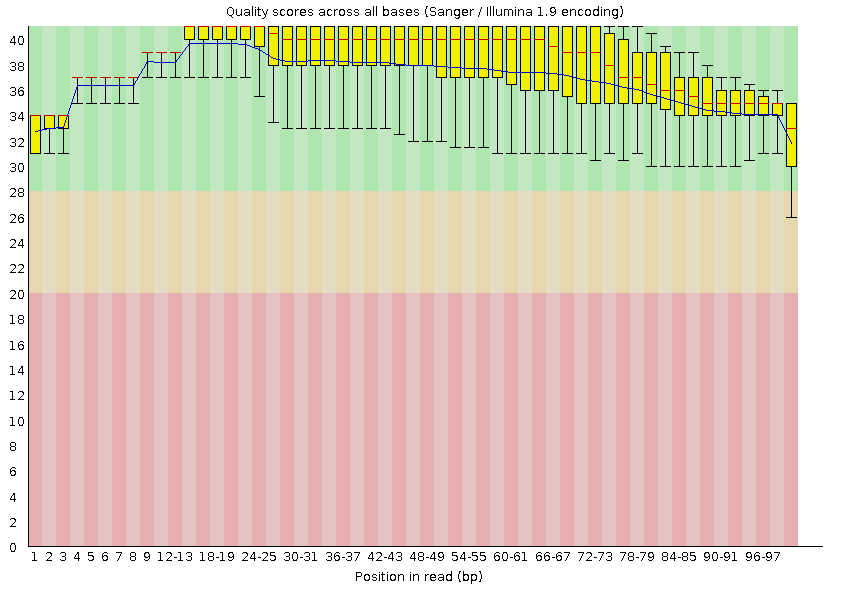
From the graph it is clear that the quality of reading the first 20 nucleotides has hardly changed. Starting at about 30 nucleotides, the scatter of values slightly decreases and the quality improves. The most noticeable improvements are noticeable for nucleotides from 68 and onwards, which was expected, because they initially had the strongest inaccuracy, which was reduced by removing nucleotides with poor quality.
Task 2. Reads mapping
Used commands are presented in Table 1. 10536 reads were unpaired, 73 of them were aligned 0 times, 10461 of them were aligned exactly one time (2 were aligned more than once).
10536 reads; of these:
10536 (100.00%) were unpaired; of these:
73 (0.69%) aligned 0 times
10461 (99.29%) aligned exactly 1 time
2 (0.02%) aligned >1 times
99.31% overall alignment rate
Task 3. SNP analysis
| Coordinate | Polymorphism type | REF/ALT | DP | Quality |
| 5113452 | SNP | C/T | 9 | 133.032 |
| 5090641 | SNP | G/A | 98 | 221.999 |
| 5122932 | SNP | A/G | 28 | 221.999 |
All in all, 106 SNPs and 5 indels have been discovered. Average quality of found SNPs is 79,6, average coverage is 12,2.
RefGene divides SNPs into four categories: intronic, exonic, intergenic, UTR3 and UTR5 (untranslated regions of mRNA 3' and 5' respectively). My rations: 78 intronic, 15 exonic, 3 intergenic and 8 UTR3. SNPs are in genes: JAK2, IL33. 2 SNPs in synonymous group, 1 in non-synonimous
From the 1000 Genomes annotation, you can get data on SNP frequencies. The average frequency is 0.507.
The highest frequency is: 0.967652 chr9 5020529 5020529 A G hom 11.3429 2
96 SNPs have rs. According to clinical annotation, patient has high risks of Crohn's disease, Endometriosis, Malaria and others.