Command table
Command table
| Command | Description |
|---|---|
| fastqc chr9.2.fastq | FastQC reads a set of sequence files and produces from each one a quality control report consisting of a number of different modules, each one of which will help to identify a different potential type of problem in your data. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9_1.fastq 9.1cut.fastq TRAILING:20 MINLEN:50 | This command cuts off parts with average quality less than 20 and removes reads that are less than 50 nucleotides long. Quality changes are presented in Fig.1 and 2 |
| hisat2-build chr9.fasta r_chr9_ind | Indexing of reference sequence |
| hisat2 -x r_chr9_ind -U 9.1cut.fastq --no-softclip -S chr9_alig.sam --summary-file hisat2.txt | Alignment of reads and reference sequence. --no-spliced-alignment was removed due to the fact that in RNA reads it is possible that 2 or more different parts of one read can be mapped distantly due to splicing. |
| samtools view -t chr9.fasta.fai -b chr9_alig.sam -o chr9_alig.bam | Conversion of alignment into binary format |
| samtools sort chr9_alig.bam -T temp -o chr9_sort.bam | Alignment sorting by the coordinate in the reference |
| samtools index chr9_sort.bam | Indexing of sorted file |
| htseq-count -f bam -s no -m intersection-nonempty -i gene_id chr9_sort.bam /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf -o file.HTSeq-count.txt | This tool takes an alignment file in SAM or BAM format and feature file in GFF format and calculates the number of reads mapping to each feature.
-f (format), --format=(format) Format of the input data. Possible values are sam (for text SAM files) and bam (for binary BAM files). Default is sam. -s (yes/no/reverse), --stranded=(yes/no/reverse) whether the data is from a strand-specific assay (default: yes) -i (id attribute), --idattr=(id attribute) GFF attribute to be used as feature ID. Several GFF lines with the same feature ID will be considered as parts of the same feature. The feature ID is used to identity the counts in the output table. The default, suitable for RNA-Seq analysis using an Ensembl GTF file, is gene_id. -m (mode), --mode=(mode) Mode to handle reads overlapping more than one feature. Possible values for (mode) are union, intersection-strict and intersection-nonempty (default: union) |
Task 1. Reads preparation
Amount of reads didn't change dramatically: from 14020 to 13487 (533 were dropped). According to the command was used, reads whose length was less than 50 (after cutting off low quality parts) were dropped.
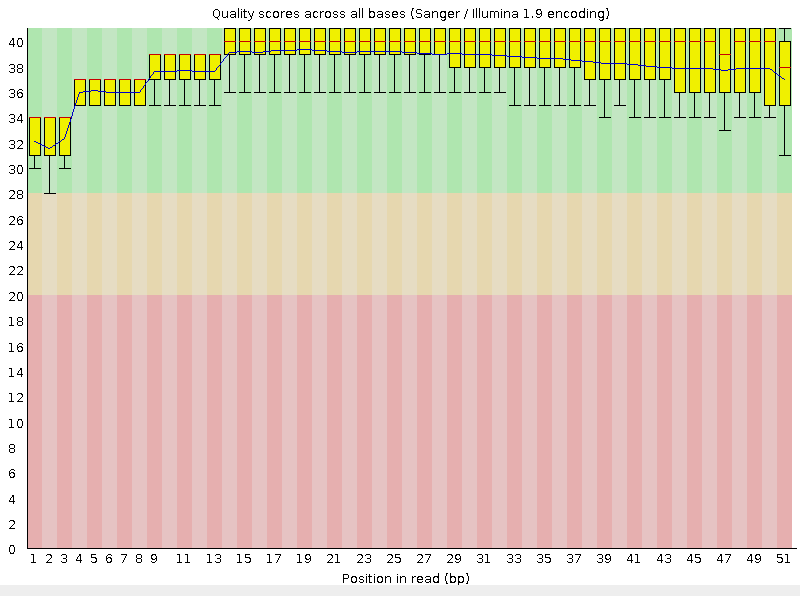

From the graph it is clear that the quality of reading the first 25 nucleotides has hardly changed. Starting at about 30th nucleotide, the scatter of values slightly decreases and the quality improves.
Task 2. Reads mapping
Used commands are presented in Table 1. 13487 reads were unpaired, 78 of them were aligned 0 times, 13409 of them were aligned exactly one time (0 were aligned more than once).
13487 reads; of these:
13487 (100.00%) were unpaired; of these:
78 (0.58%) aligned 0 times
13409 (99.42%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.42% overall alignment rate
Task 3. Reads count
Results of count.py script: __no_feature - 251; ENSG00000119335.12 gene - 13158;__not_aligned - 78
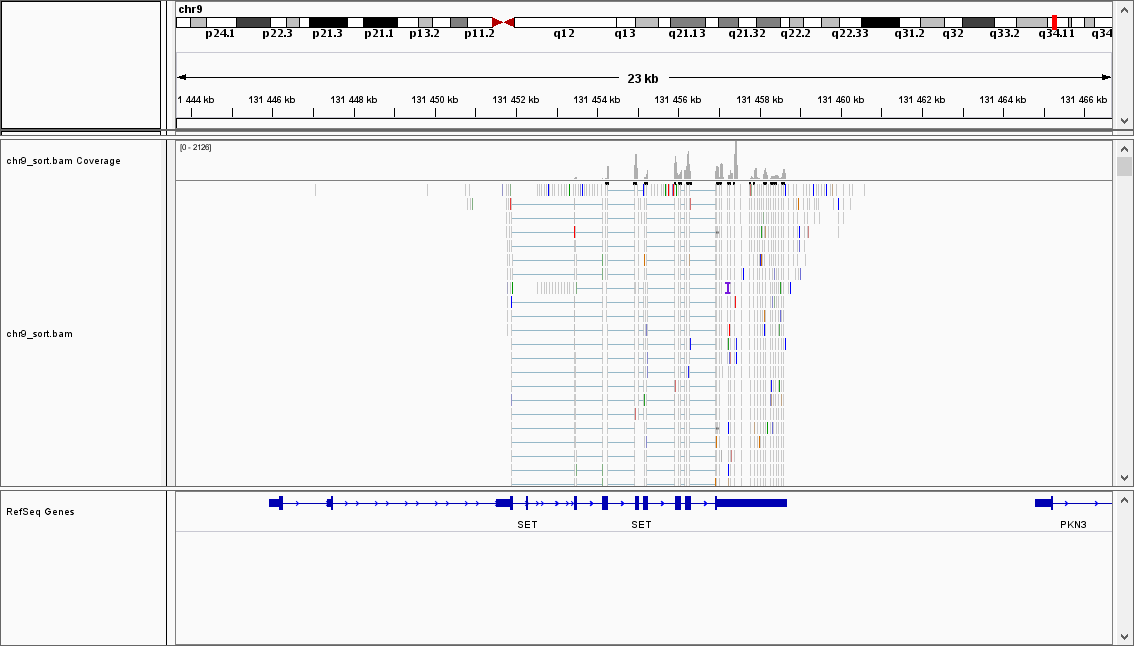
ENSG00000119335.12 is SET-201 gene, coding Protein SET. Multitasking protein, involved in apoptosis, transcription, nucleosome assembly and histone chaperoning.
It also seems that all reads ended up whithin ENSG00000119335.12 gene