>gene argB E.coli atgatgaatccattaattatcaaactgggcggcgtactgctggatagtgaagaggcgctg gaacgtctgtttagcgcactggtgaattatcgtgagtcacatcagcgtccgctggtgatt gtgcacggcggcggttgcgtggtggatgagctgatgaaagggctgaatctgccggtgaaa aagaaaaacggcctgcgggtgacgcctgctgatcagatagacattatcaccggagcactg gcgggaacggcaaataaaaccctgttggcatgggcgaagaaacatcagattgcggccgta ggtttgtttctcggtgacggcgacagcgtcaaagtgacccagcttgatgaagagttaggt catgttggactggcgcagccaggttcgcctaagcttatcaactccttgctggagaacggt tatctgccggtggtcagctccattggcgtaacagacgaagggcaactgatgaacgtcaat gccgaccaggcggcaacggcgctggcggcaacgctgggcgcggatctgattttgctctcc gacgtcagcggcattctcgacggcaaagggcaacgcattgccgaaatgaccgccgcgaaa gcagaacaactgattgagcagggcattattactgacggcatgatagtgaaagtgaacgcg gcgctggatgcggcccgcacgctgggccgtccggtagatatcgcctcctggcgtcatgcg gagcagcttccggcactgtttaacggtatgccgatgggtacgcggattttagcttaa
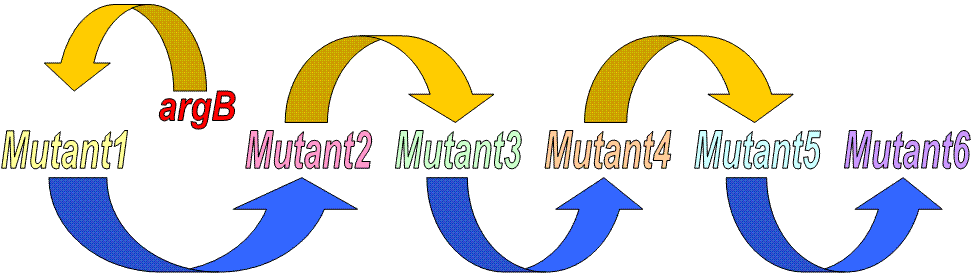
этап эволюции "истинные" расстояния (число замен на 100 нуклеотидов) общее число замен (длина гена argB = 777 п.н.) gene argB__Mutant1 10 ~78 Mutant1__Mutant2 10 ~78 Mutant2__Mutant3 30 ~233 Mutant3__Mutant4 25 ~194 Mutant4__Mutant5 50 ~389 Mutant5__Mutant6 50 ~389 |
Данную таблицу удобнее преобразовать следующим образом:
gene argB 0 10 20 50 75 125 175
Mutant1 0 10 40 65 115 165
Mutant2 0 30 55 105 155
Mutant3 0 25 75 125
Mutant4 0 50 100
Mutant5 0 50
Mutant6 0
gene argB Mutant1 Mutant2 Mutant3 Mutant4 Mutant5 Mutant6
|
В каждой ячейке данной таблицы стоят "истинные" попарные расстояния между последовательностями гена и мутантами. Причем расстояния между "дальними" мутантами - между ними произошло не менее 2-х эволюционных событий - можно рассчитать по следующей формуле (также если приписать исходному гену название "Mutant0":
Nij = ∑Nk-1,k
где Nij - эволюционное расстояние между генами i и j; суммирование в пределах от i+1 до j.
msbar argB.fasta m1.fasta -point 4 -count 84 -auto
данная строка расшифровывается так: последовательность argB.fasta изменяется случайным образом в последовательность m1.fasta посредством введения 84 замен (count 4).
|
msbar argb.fasta m1.fasta -point 4 -count 78 -auto; msbar m1.fasta m2.fasta -point 4 -count 78 -auto; msbar m2.fasta m3.fasta -point 4 -count 233 -auto; msbar m3.fasta m4.fasta -point 4 -count 194 -auto; msbar m4.fasta m5.fasta -point 4 -count 389 -auto; msbar m5.fasta m6.fasta -point 4 -count 389 -auto; cat m1.fasta >> argb.fasta; cat m2.fasta >> argb.fasta; cat m3.fasta >> argb.fasta; cat m4.fasta >> argb.fasta; cat m5.fasta >> argb.fasta; cat m6.fasta >> argb.fasta |
Чтобы исполнить его, использовались команды:
chmod +x my.script
./my.script
Для этого, в строке Unix вводилась следующая команда:
distmat -sequence argB.fasta -outfile D.distmat -nucmethod 0 - в файле D.distmat представлена матрица попарных различий.
distmat -sequence argB.fasta -outfile JC.distmat -nucmethod 1 - в файле JC.distmat представлена матрица попарных расстояний, вычисленных по алгоритму Джукса-Кантора.
Для их представления в более читабельном виде и сравнения создана книга Exel - evol, в которой на странице All_data представлены все три матрицы, а на странице Comparison они подвергнуты сравнению. В результате сравнения получен следующий график (который, правда несколько оптимизирован и увеличен, поэтому представлен в книге на странице GRAPH):
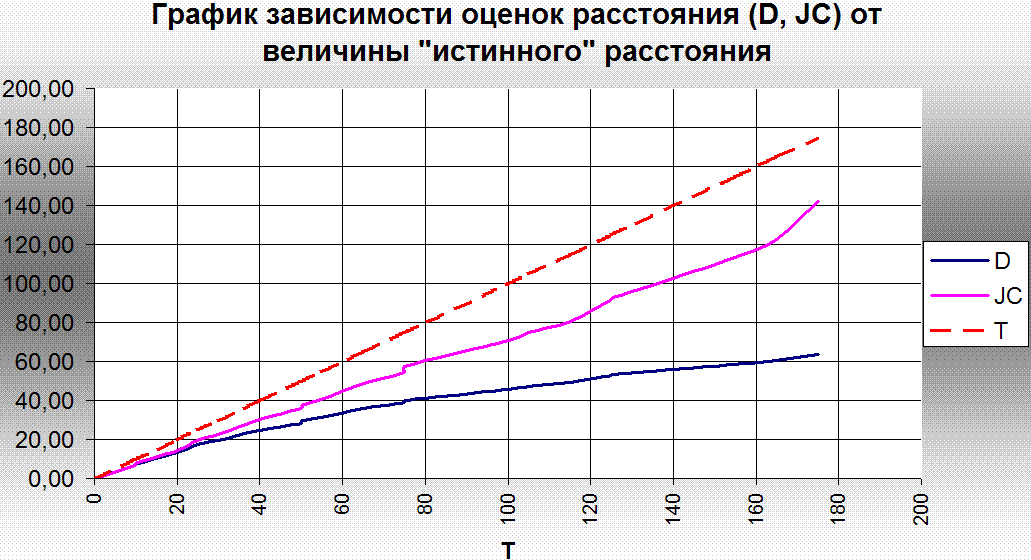
Рис.2. График оценок и истинных расстояний.
На рисунке красной пунктирной линией показаны величины "истинных" эволюционных попарных расстояний, синей сплошной - величины попарных различий, розовой сплошной - величины попарных расстояний, вычисленных по алгоритму Джукса-Кантора. В подписях к оси X кроме значения "истинных" расстояний, также приведены обозначения сравниваемых попарно последовательностей: g - значит последовательность гена argB; mX - последовательность мутанта mX. Тогда обозначение "mXmY - попарное сравнение последовательностей мутанта mX с мутантом mY.
Итак, для "истинных" расстояний учитывается каждая нуклеотидная мутация. Таким образом, график зависимости числа замен от истинных замен представляет собой прямую пропорциональность. При малом числе мутаций, что приблизительно составляет 15 нуклеотидных замен на 100 нуклеотидов, все три графика (прямая истинных расстояний (T), оценки по Джуксу-Кантору (JC) и графика попарных различий (D)) практически идентичны. А вот дальше отличия проявляются и весьма заметные.
Кривая попарных различий расположена ниже графика "истинных расстояний". При этом видно, что она постепенно стремится к горизонтальной прямой - ассимптоте: возможно, приблизительно, эта прямая составляет 70-80 нуклеотидных замен на 100 нуклеотидов., так как кривая, впринципе, пересекает значение в 60 нуклеотидных замен на 100 нуклеотидов последовательности и далее некоторым образом растет, но очевидно (так как производная графика довольно мала [так "на глазок"]) она приближается к определенной горизонтальной ассимптоте. Видимо, это связано с инструментальной ошибкой метода оценивания, так как при данном способе оценки не учитываются возможные обратные замены, отчего при большом числе замен, общее количесто различных замен уменьшается, и относительный "рост" графика замедляется: он выходит на плато.
Также, таким метод сравнения двух последовательностей не учитываются последовательные различные мутации в одном кодоне, то есть, например, процесс изменения ТТТ в исходной последовательности >> ТТС в промежуточной последовательности >> ТТА в результарующей последовательности приводят к идентификации одного различия начального и конечного кодонов, в то время как произошло 2 мутации. Кривая, построенная на основании данных расчета по алгоритму Джукс – Кантора (модель предполагает равные вероятности замен между всеми четырьмя типами нуклеотидов с вероятностью р = a), также лежит несколько ниже графика "истинных расстояний", но выше кривой для попарных различий на 100 нуклеотидов. Можно отметить, что при числе истинных замен, равном 175, число замен, рассчитанных по методу Джукса-Кантора, составляет около 142, то есть расчетная величина отличается от реальной на: (175 - 142)/175 * 100% = 18,9%, тогда как расчетная величина, полученная от использования метода попарных различий, отличается от реальной на целых (175 - 64)/175 * 100% = 63,4%!!!. Фактически, метод оценки эволюционных расстояний по алгоритму Джукса-Кантора эффуктивнее на целых 44,5%!!, чем алгоритм оцеки эволюционных расстояний по методу попарных различий.
Очевидно, это связано с тем, что алгоритм Джукса-Кантора учитывает вероятность возможных обратных замен, тогда как метод оценки попарных различий совершенно не способен к такому учету.