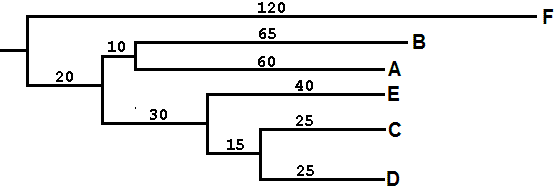
(((А:60,В:65):10,((С:25,D:25):15,Е:40):30):20,F:120);
|
A B C D E F
* * . . * *
* * . . . *
* * . . . .
|
|
N = 777/100*n = 7,77*n
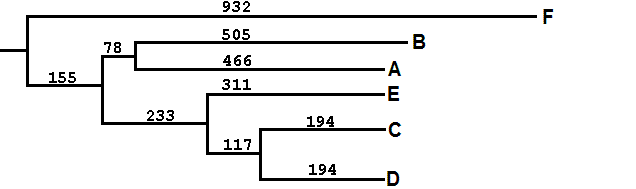 |
msbar argb.fasta mABCDEF.fasta -point 4 -count 155 -auto msbar argb.fasta mF.fasta -point 4 -count 932 -auto msbar mABCDEF.fasta mAB.fasta -point 4 -count 78 -auto msbar mABCDEF.fasta mCDE.fasta -point 4 -count 233 -auto msbar mAB.fasta mA.fasta -point 4 -count 466 -auto msbar mAB.fasta mB.fasta -point 4 -count 505 -auto msbar mCDE.fasta mCD.fasta -point 4 -count 117 -auto msbar mCDE.fasta mE.fasta -point 4 -count 311 -auto msbar mCD.fasta mC.fasta -point 4 -count 194 -auto msbar mCD.fasta mD.fasta -point 4 -count 194 -auto cat mF.fasta >> argbz.fasta cat mA.fasta >> argbz.fasta cat mB.fasta >> argbz.fasta cat mC.fasta >> argbz.fasta cat mD.fasta >> argbz.fasta cat mE.fasta >> argbz.fasta |
fdnaml argbz.fasta -ttratio 1 -auto
+--------mE.
+------4
| | +-----mD.
| +--3
| +-----mC.
|
| +--------------mA.
2---1
| +---------------mB.
|
+------------------------------------mF.
|
fdnadist argbz.fasta -ttratio 1 -auto
Программа fdnadish использует множественное выравнивание последовательностей для подсчета попарных расстояний. Но так как работа проводится в предположении, что эволюция происходит путем точечных замен (без учета возможных инверсий, транспозиций, кроссинговера и др.), то выравнивание которое, нужно подавать на вход программе, совпадает с содержимым файла argbz.fasta. Подсчеты программы сохранены в файле argbz.fdnadist. В этой матрице расстояний эволюционные расстояния приведены в количестве замен, приходящемся на 1 н.п (а не на 100 н.п.).
В более удобном виде матрица попарных расстояний представлена ниже (расстояния переведены в количество замен на 100 нуклеотидов):
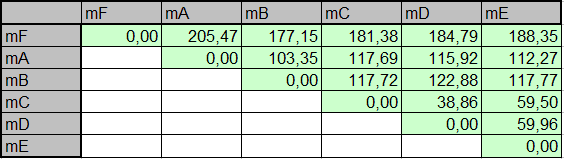
Вычисленные расстояния в целом соответствуют истинным эволюционным расстояниям, но все же есть некоторые различия. Так наибольшие различия (до 200 замен на 100 н.п.) свойственны последовательностям mutF и mutA, хотя в истинном дереве наиболее удаленными являются мутанты mutB и mutF (теоретически, расстояние между ними равно 65 + 10 + 20 + 120 = 215 нуклеотидных замен), хотя для них рассчитано 177 нуклеотидных замен. Видимо это является следствием инструментальной ошибки алгоритма джукса-Кантора, проявляетмой при достаточно высоких эволюционных расстояний.
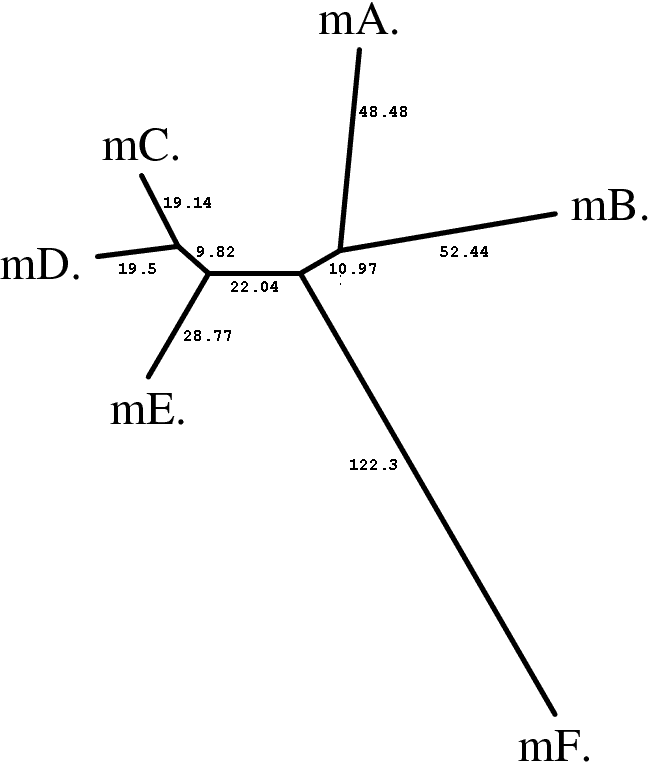
Для получения дерева с помощью алгоритма Neighbor-joining, этот файл подавался на вход программе fneighbor, используя следующую команду :
fneighbor argbz.fdnadist -auto
+----------------mA.
+--3
! +-------------mB.
!
! +-----mC.
! +---1
4-------2 +-----mD.
! !
! +--------mE.
!
+--------------------------------------mF.
|
fneighbor argbz.fdnadist -treetype u -auto
+-------------------------------------------------------mF.
!
--5 +------------------------------mA.
! +---3
! ! +------------------------------mB.
+--------------------4
! +----------mC.
! +------1
+----------------2 +----------mD.
!
+-----------------mE.
|
тип ветви: A B C D E F |
True | Maximum likelihood | Neighbor-joining | UPGMA |
* * . . * * |
X | X | X | X |
* * . . . * |
X | X | X | X |
. . . . . * |
X | X | X | X |
. . * * * * |
X | X | X | X |
 |
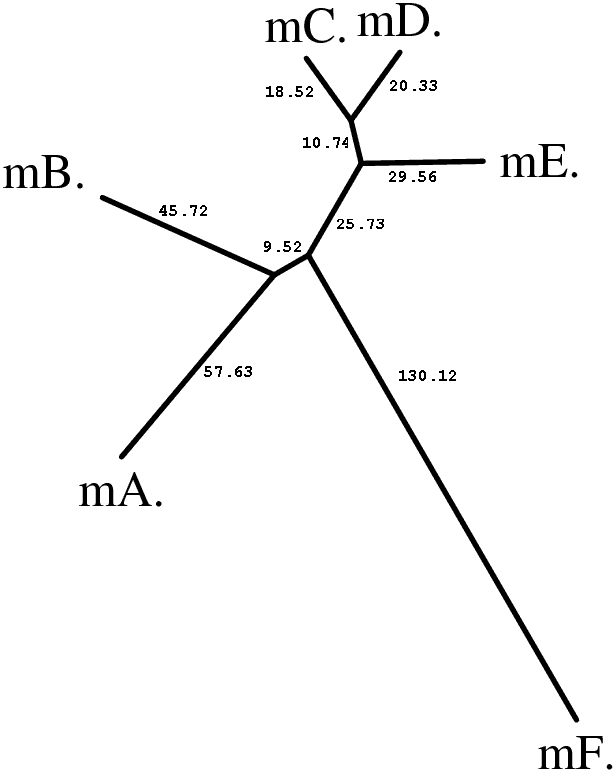 |
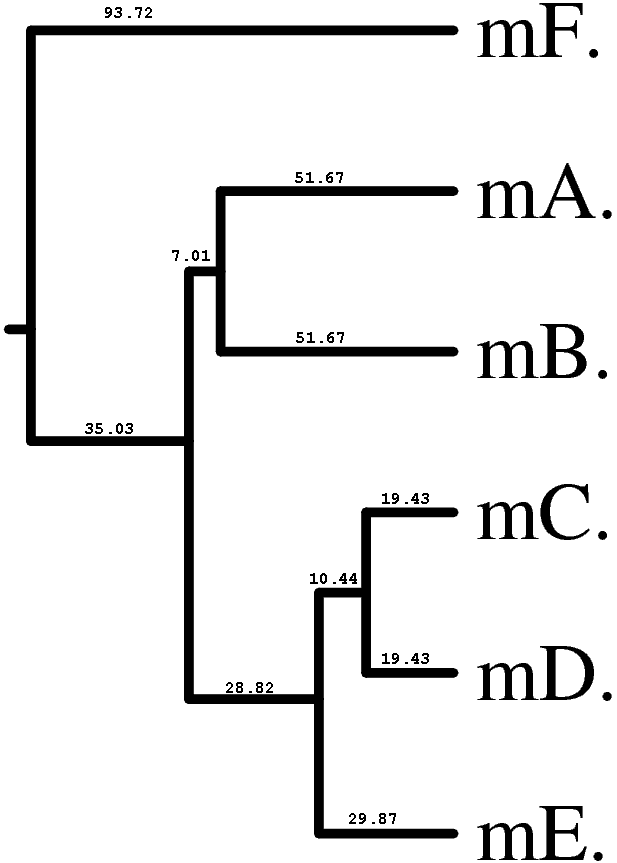 |
|
S = [1/(n - 1)*Σ(Xi - Xp)2]
программа |
NJ | ML | UPGMA |
оценка |
9.25 | 10.52 | 22.26 |
fseqboot argbz.fasta -auto
fconsense argb_100.treefile -auto
Extended majority rule consensus tree (метод построения (по умолчанию)
CONSENSUS TREE:
the numbers on the branches indicate the number
of times the partition of the species into the two sets
which are separated by that branch occurred
among the trees, out of 100.00 trees
+------mD.
+-96.0-|
+-95.0-| +------mC.
| |
+-66.0-| +-------------mE.
| |
+------| +--------------------mF.
| |
| +---------------------------mA.
|
+----------------------------------mB.
|
Species in order (мутантные последвательности, соответствующие листьям): 1. mB. 2. mA. 3. mE. 4. mD. 5. mC. 6. mF. Sets included in the consensus tree (ветви, включенные в консенсусное дерево): Set (species in order) How many times out of 100.00 (частота встречаемости) ...**. 96.00 ..***. 95.00 ..**** 66.00 Sets NOT included in consensus tree (ветви, не включенные в консенсусное дерево): Set (species in order) How many times out of 100.00 .****. 32.00 ...*** 5.00 ....** 3.00 .*...* 2.00 ...*.* 1.00 |
|
тип ветви: A B C D E F |
реальное дерево | реконструированное дерево |
* * . . * * |
X | X |
* * . . . * |
X | X |
. . . . . * |
X | |
. . * * * * |
X | X |
fdrawtree tree.txt tree.ps -auto
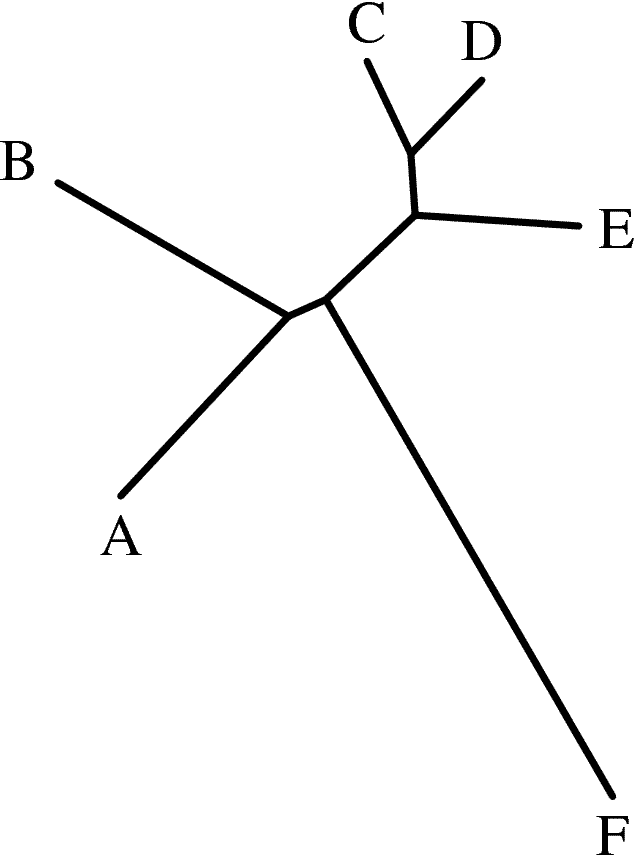 |