fseqboot argbz.fasta -test j
fdnaml jackknife.fseqboot -ttratio 1 -auto
fconsense jackknife.treefile -auto
CONSENSUS TREE:
the numbers on the branches indicate the number
of times the partition of the species into the two sets
which are separated by that branch occurred
among the trees, out of 100.00 trees
(числа, расположенные на ветвях, обозначают количество
деревьев, в которых данная ветвь встретилась, в расчете
на 100 филогенетических деревьев (размер выборки).
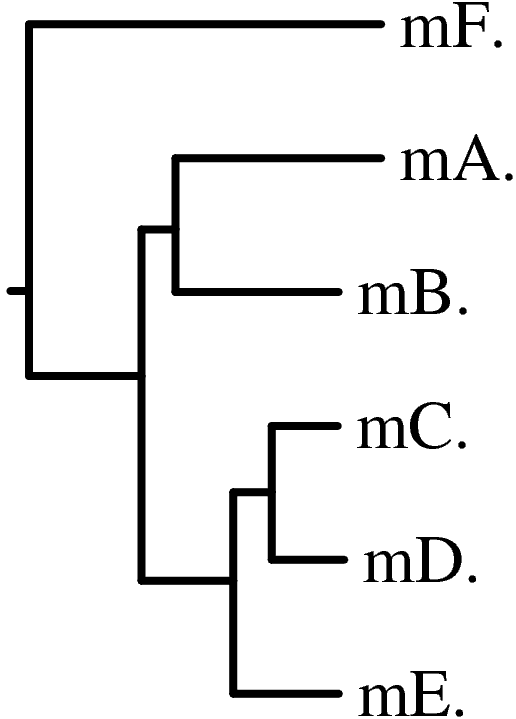
+------mD.
+-------100.0-|
| +------mC.
+------|
| | +------mB.
| | +-98.0-|
| +100.0-| +------mA.
| |
| +-------------mF.
|
+---------------------------mE.
|
+------mD.
+-------100.0-|
| +------mC.
+------|
| | +------mB.
| | +-98.0-|
| +100.0-| +------mA.
| |
| +-------------mF.
|
+---------------------------mE.
|
+------mD.
+-96.0-|
+-95.0-| +------mC.
| |
+-66.0-| +-------------mE.
| |
+------| +--------------------mF.
| |
| +---------------------------mA.
|
+----------------------------------mB.
|
Порядок следования листьев в формулах
разбиения множества ветвей.
1. mE.
2. mD.
3. mC.
4. mB.
5. mA.
6. mF.
Ветви, включенные в дерево:
Тип ветви частота встречаемости ветви в
выборке деревьев
...*** 100.00
.**... 100.00
...**. 98.00
Ветви, не включенные в дерево:
Тип ветви частота встречаемости ветви в
выборке деревьев
...*.* 2.00
|
Порядок следования листьев в формулах
разбиения множества ветвей. (Изначально
он был иной, но для удобства сравнения
изменен)
1. mE.
2. mD.
3. mC.
4. mB.
5. mA.
6. mF.
Ветви, включенные в дерево:
Тип ветви частота встречаемости ветви в
выборке деревьев
...*** 96.00
**.... 95.00
***..* 66.00
Ветви, не включенные в дерево:
Тип ветви частота встречаемости ветви в
выборке деревьев
***.*. 32.00
*.*..* 5.00
..*..* 3.00
.*...* 2.00
*....* 1.00
|
eff = Σ xi / [n*100] * 100%
| метод оценки | jackknife | bootstrap |
| эффективность | 98.7% | 42.7% |
fretree 6 argbz_fneighbor.treefile
fdrawgram fretree.ps fretree1.ps -style p
|
fdnamlk argbz.fasta fretree.ps argbz.fdnamlk -ttratio 1 -hypstate Y
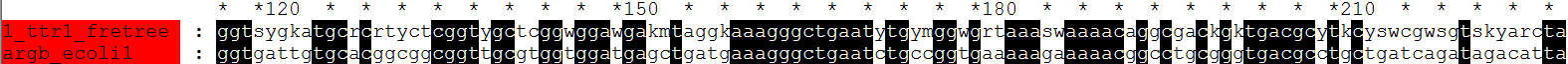 |
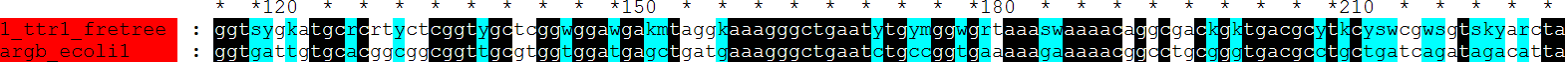 |
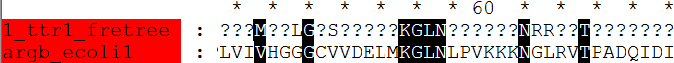 |
fdnamlk argbz.fasta fretree.ps argbzm.fdnamlk -ttratio n -hypstate Y
 |
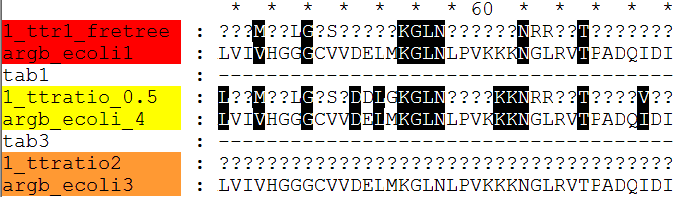 |
fdnamlk argbz.fasta argbz_fdnaml.treefile argbz8.fdnamlk -freqsfrom N -basefreq 0.25_0.25_0.25_0.25 -ttratio 0.5 -hypstate Y
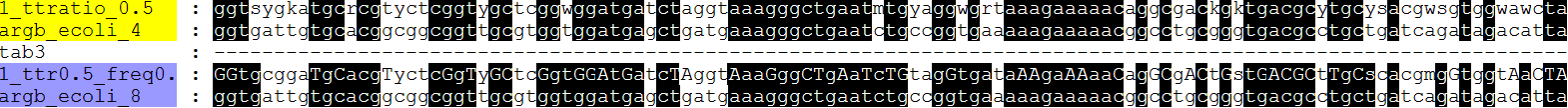 |