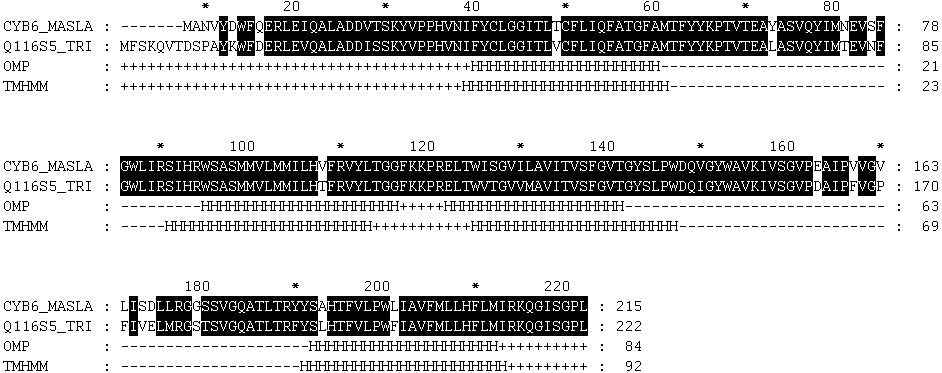
CYB6_MASLA 1 MANVYDWFQERLEIQALADDVTSKYVPPHVNIFYCLGGITLTCFLIQFAT 50
||||||||||||||||||||||||||||||||||||||||||||||||||
SEQUENCE 1 MANVYDWFQERLEIQALADDVTSKYVPPHVNIFYCLGGITLTCFLIQFAT 50
CYB6_MASLA 51 GFAMTFYYKPTVTEAYASVQYIMNEVSFGWLIRSIHRWSASMMVLMMILH 100
||||||||||||||||||||||||||||||||||||||||||||||||||
SEQUENCE 51 GFAMTFYYKPTVTEAYASVQYIMNEVSFGWLIRSIHRWSASMMVLMMILH 100
CYB6_MASLA 101 VFRVYLTGGFKKPRELTWISGVILAVITVSFGVTGYSLPWDQVGYWAVKI 150
||||||||||||||||||||||||||||||||||||||||||||||||||
SEQUENCE 101 VFRVYLTGGFKKPRELTWISGVILAVITVSFGVTGYSLPWDQVGYWAVKI 150
CYB6_MASLA 151 VSGVPEAIPVVGVLISDLLRGGSSVGQATLTRYYSAHTFVLPWLIAVFML 200
||||||||||||||||||||||||||||||||||||||||||||||||||
SEQUENCE 151 VSGVPEAIPVVGVLISDLLRGGSSVGQATLTRYYSAHTFVLPWLIAVFML 200
CYB6_MASLA 201 LHFLMIRKQGISGPL 215
|||||||||||||||
SEQUENCE 201 LHFLMIRKQGISGPL 215
|
#=======================================
#
# Aligned_sequences: 2
# 1: CYB6_MASLA
# 2: Q116S5_TRIEI
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 222
# Identity (ID): 184/222 (82.9%)
# Similarity: 199/222 (89.6%)
# Gaps: 7/222 ( 3.2%)
# Score: 1001.0
#
#
#=======================================
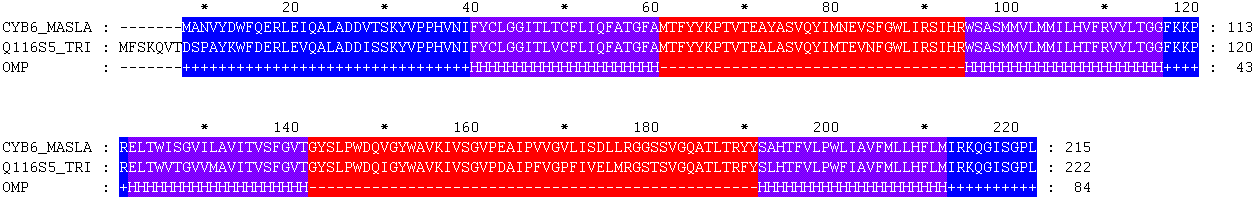
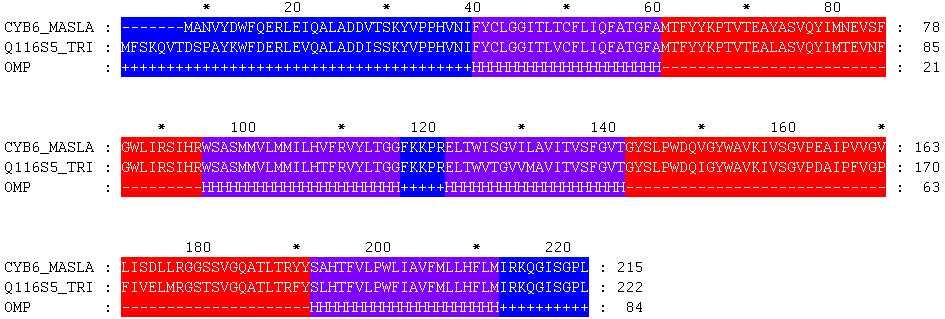
CYB6_MASLA 1 -------MANVYDWFQERLEIQALADDVTSKYVPPHVNIFYCLGGITLTC 43
.:..|.||.||||:||||||::|||||||||||||||||||.|
Q116S5_TRIEI 1 MFSKQVTDSPAYKWFDERLEVQALADDISSKYVPPHVNIFYCLGGITLVC 50
CYB6_MASLA 44 FLIQFATGFAMTFYYKPTVTEAYASVQYIMNEVSFGWLIRSIHRWSASMM 93
||||||||||||||||||||||.|||||||.||:||||||||||||||||
Q116S5_TRIEI 51 FLIQFATGFAMTFYYKPTVTEALASVQYIMTEVNFGWLIRSIHRWSASMM 100
CYB6_MASLA 94 VLMMILHVFRVYLTGGFKKPRELTWISGVILAVITVSFGVTGYSLPWDQV 143
|||||||.|||||||||||||||||::||::||||||||||||||||||:
Q116S5_TRIEI 101 VLMMILHTFRVYLTGGFKKPRELTWVTGVVMAVITVSFGVTGYSLPWDQI 150
CYB6_MASLA 144 GYWAVKIVSGVPEAIPVVGVLISDLLRGGSSVGQATLTRYYSAHTFVLPW 193
||||||||||||:|||.||..|.:|:||.:|||||||||:||.|||||||
Q116S5_TRIEI 151 GYWAVKIVSGVPDAIPFVGPFIVELMRGSTSVGQATLTRFYSLHTFVLPW 200
CYB6_MASLA 194 LIAVFMLLHFLMIRKQGISGPL 215
.|||||||||||||||||||||
Q116S5_TRIEI 201 FIAVFMLLHFLMIRKQGISGPL 222
|
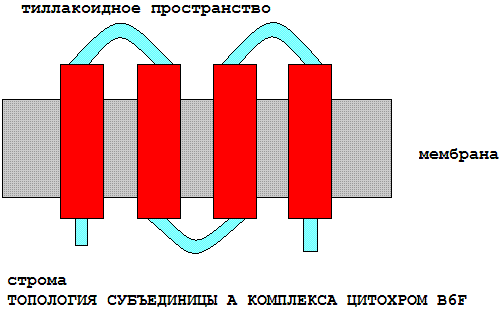
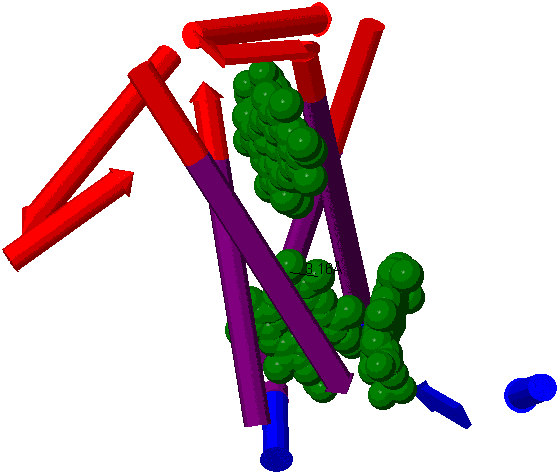
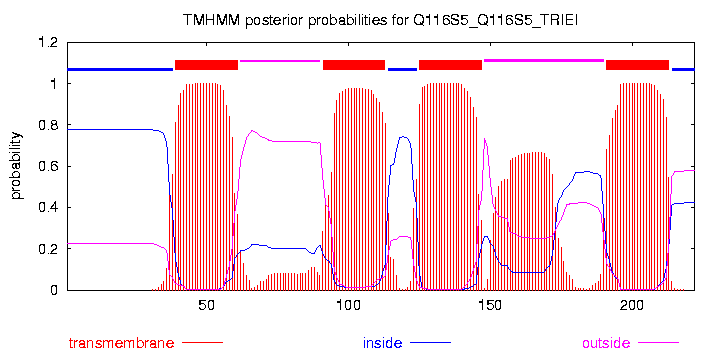
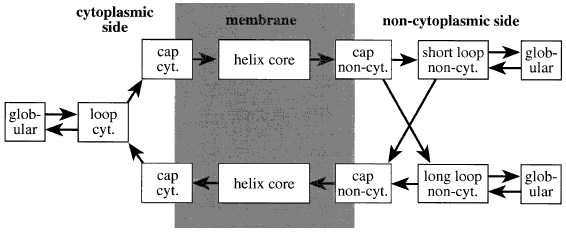
| Элемент вторичной структуры | начало | конец |
| стромальная (внутренняя) петля | 1 | 38 |
| трансмембранная спираль | 39 | 61 |
| тиллакоидная (внешняя) петля | 62 | 90 |
| трансмембранная спираль | 91 | 113 |
| стромальная (внутренняя) петля | 114 | 124 |
| трансмембранная спираль | 125 | 147 |
| тиллакоидная (внешняя) петля | 148 | 190 |
| трансмембранная спираль | 191 | 213 |
| стромальная (внутренняя) петля | 214 | 215 |
| Число аминокислотных остатков (или доля а.о.) | |
| Всего а.к. остатков | 222 |
| Остатки, предсказанные как локализованные в мембране (всего) | 92 |
| Правильно предсказано (true positives, TP) | 78 |
| Предсказано не то, что нужно (а.о. предсказаны как мембранные, а по данным ОРМ таковыми не являются, false positives, FP) | 14 |
| Правильно не предсказано ( не предсказаны, и по данным ОРМ не находятся в мембране, true negatives, TN) | 124 |
| Не предсказано то, что нужно (остатки по данным ОРМ находятся в мембране, false negatives, FN) | 6 |
| Чувствительность (sensivity) = TP / (TP+FN) | 0,93 |
| Специфичность (specificity) = TN / (TN+FP) | 0,90 |
| Точность (precision) = TP / (TP+FP) | 0,85 |
| Сверхпредсказание = FP/ (FP+TP) | 0,15 |
| Недопредсказание = FN / (TN+FN) | 0,05 |