На главную страницу второго семестра.
"Филогенетические деревья, реконструированные разными способами".
I. Построение дерева по алгоритму UPGMA.
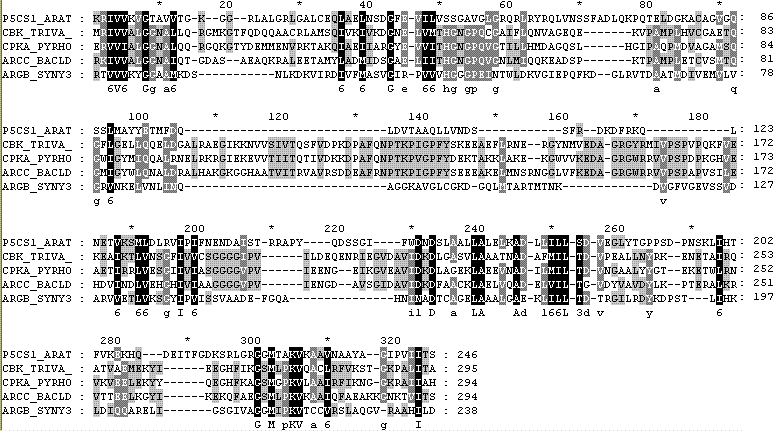
Для построения дерева использовались последовательности пяти белков, содержащих домен AA_kinase. На основе последовательностей домена пяти белков: CBK_TRIVA, CPKA_PYRHO, ARCC_BACLD, ARGB_SYNY3, P5CS1_ARATH построено эталонное выравнивание, для которого рассчитана матрица попарных совпадений.
Рис.1. Эталонное выравнивание, полученное из БД PFAM с описанием семейства белков AA_kinase.
Рис.2. Матрица попарных совпадений.
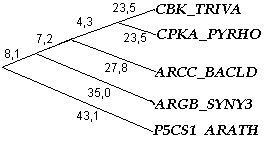
Результаты выполнения упражнения можно увидеть в книге UPGMA.xls, а также на рисунке ниже приведено построенное дерево:
Рис.3. Филогенетическое дерево для последовательностей домена AA_kinase в пяти белках, построенное с помощью алгоритма UPGMA (сделано вручную).
Для вышеприведенного дерева скобочная формула выглядит следующим образом: ((((CBK_TRIVA:23.5, CPKA_PYRHO:23.5):4.3, ARCC_BACLD:27.8):7.2, ARGB_SYNY3:35.0):8.1, P5CS1_ARATH:43.1);
II. Получение и cравнение 2-ух изображений построенного дерева.
При выполнении задания использовались программы визуализации дерева drawtree и drawgram. В обеих программах на вход подавалась правильная скобочная формула, и по прошествии некоторого времени, результат представлялся в файлах ps-формата (их можно взять здесь и здесь).
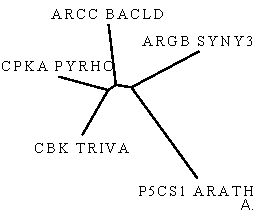
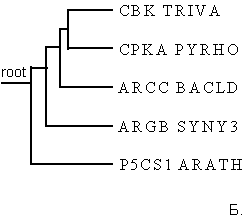
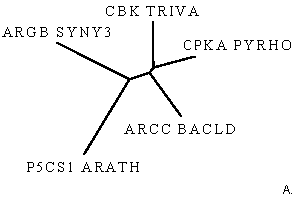
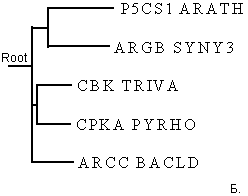
Рис.4.Филогенетические деревья для последовательностей домена AA_kinase в пяти белках, визуализированные программами drawtree (рис.4А) и drawgram (рис.4Б).
Заметно, что реализуя алгоритм UPGMA, мы всегда получаем ультраметрическое дерево. Если мы верим данным результатам, то мы допускаем, что для эволюции данных последовательностей справедлива теория "молекулярных часов": за равное время во всех ветвях эволюции накапливается равное число мутаций. Так как задача сравнить изображения двух деревьев, то примем эту теорию (пока) и не будем вдаваться глубоко в дебри. Также заметно, что на рис.4А изображено неукорененное ультраметрическое дерево с различными длинами ветвей (указана не только топология дерева), а на рис.4Б приведено укорененное ультраметрическое дерево в виде кладограммы. На мой взгляд, более достоверным результатом стоит считать неукорененное дерево на рис.4А, так как предполагать нахождение узла предковой последовательности в какой-то зафиксированной точке не весьма корректно. В принципе, предковая последовательность может находиться на любой ветви, хоть даже между листом и другими ветвями, главное, чтобы такое укоренное дерево правильно отражало историю эволюции интересующего предмета. Также выборка белков очень маленькая (всего пять последовательностей) и располагать корень где-либо очень опасно (как выяснилось, просто по умолчанию в программе в поле "Ancestral nodes" стояло значение "Intermediate" - промежуточный, отчего на выходе получалось укорененное дерево. Поэтому я полагаю лучше доверять изображению рис.4А, отражающее топологию ветвей и листьев. Также я доверяю ему по следующим причинам. Из анализа аннотаций данных последовательностей можно изобразить неукорененное дерево как на рис4А, но вместо подписей с названиями последовательностей белков, поставим таксоны организмов, из которых они были секвенированы. Тогда получим следующий рисунок:
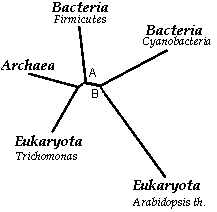
Рис.5.Филогенетические дерево для последовательностей домена AA_kinase в пяти белках, с отмеченной таксономией организмов, содержащих эти белки.
По этому дереву видно, что если строить эволюцию, нужно очень сильно извертеться, чтобы не запутаться, где все же определить узел предковой последовательности (а не ставить на любом месте, как это делает программа). Судя по белкам и эволюционным расстояниям, наиболее близкими последовательностями оказались из Archea и Eucaryota, Trichomonas. Для меня это довольно ошеломляющий результат. Ведь по определению одного из биологов, архейные, это "бактерии по форме, и эукариоты по содержанию": "форма бактерий", значит нет ядра и цитоскелета, одноклеточны и малы по размерам, но в геноме довольно много и эукариотических генов ("эукариоты по содержанию"). Таким образом, если предполагать, что CPKA_PYRHO (Archea) и CBK_TRIVA (Eucaryota) разошлись за одно событие от одного предка, то очень нужно постараться представить себе это событие, что от какого-то "монстра-предка" вдруг произошел эукариотический белок в трихомонасе и белок в составе протеома архейного организма. Конечно, можно сослаться на скромные размеры выборки белков, но это будет не этично в плане задания: попытаться описать эволюцию последовательностей. Другая замеченная особенность: расхождение цианобактерий (последовательность ARGB_SYNY3), которых также относят к низшим водорослям, рассматривая в курсе альгологии, и высших наземных растений (последовательность P5CS1_ARATH) от некоторого гипотетичного предка, что вполне оправданно. Ну и остается ещё один узел предковой последовательности, ведущей к последовательности бактериальоного белка ARCC_BACLD (Bacteria). Но так как дерево не укорененное, то сказать, откуда произошли эти три предковые последовательности, предваряющие эволюцию белков с этим доменом в трех царствах: Растений, Животных, Бактерий, - просто невозможно. В целом, я предлагаю следующий механизм возможного способа описания эволюции, но сперва, чтобы было вообще хорошо, доопределим узел предковой последовательности на ветви AB (помеченной на рис.5). Тогда получим рис.6:
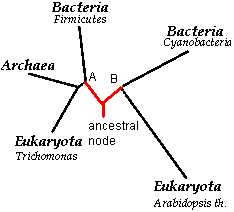
Рис.6."Дополненное филогенетическое дерево".
Тогда, допустим, что от некоторого предка (ancestral node), [возможно его содержал тот самый организм из "первичного бульона"] произошли две последовательности белков в составе двух микроорганизмов (ортологи). На данном этапе эволюция последовательностей разделяется на развитие в животных организмах (белок А) и развитие в фотосинтетиках (белок В) [что кажется правдоподобным]. Тогда от белка В, накапливая определенные мутации и подвергаясь движущему отбору, эволюционирует ARGB_SYNY3 (в составе цианобактерий), а также, в ходе более длительного времени (видно, что эволюционное расстояние больше), образуется P5CS1_ARATH в составе высших растений (видимо на одной из стадий на этой ветке происходит то, что любят рассказывать в школе (одна из баек!!): одна из промежуточных форм (уже эукариотический организм!) заглатывает цианобактерию, отчего у неё появляется новая органелла - хлоропласт, отчего животное способно к фотосинтезу. Таким образом понятно аличие узла между ARGB_SYNY3 (Cyanobacteria), P5CS1_ARATH (Arabidopsis th.). Проследим эволюцию от узла А. Видимо, белок А находился в составе протеома прокариотического организма, так как от него развивается ARCC_BACLD (Bacteria Firmicutes - бацилла), видимо в ходе дальнейшей специализации на безъядерном состоянии, и новая переходная форма, давшая начало архейному белку CPKA_PYRHO и эукариотическому CBK_TRIVA. Но учитывая явную близость последовательностей CBK_TRIVA и CPKA_PYRHO (все же в ходе алгоритма образуют кластер!!), я доверяю несколько иному сценарию. Я полагаю, что новая переходная форма представляла собой последовательность белка в составе архейного организма. Так как геном организован в плазмидах, способных к транспозиции между организмами в ходе конъюгации, то можно предположить, что на определенной стадии происходит событие: архея, перепутав организм для миксинга (обмена генами), конъюгирует с заблудшей эукариотой и происходит горизонтальный перенос генов в составе плазмиды от археи к эукариоте, и среди них был ген, кодировавший домен AA_kinase!! Кстати, эти две последовательности: CBK_TRIVA (eucaryota) и CPKA_PYRHO (Archea), можно обозвать одним "нехорошим" словом - ксенологами. Тогда в ходе дальнейшей эволюции "новой переходной формы" в составе археи образуется белок CPKA_PYRHO (Archea), а эукариота, получив и закрепив в своем геноме последовательность плазмиды, развила из последовательности гена, кодирующего домен AA_kinase, последовательность гена нового белка: CBK_TRIVA (eucaryota trichomonas).
III. Получение и описание дерева, построенного по методу ближайших соседей.
Для выполнения задания использовались те же программы для визуализации дерева: drawtree и drawgram, но на вход им подвалась скобочная формула дерева, построенного на основе множественного выравнивания. Результаты представлены на рис.7:
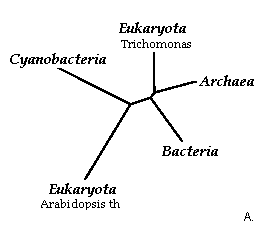
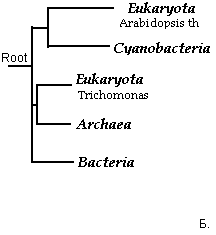
Рис.7."Дополненное филогенетическое дерево".
Да, вообщем, как и следовало ожидать, с использованием метода объединения соседей результат даже для укорененного дерева получился такой, про который я столь долго распинался выше (даже узел предковой последовательности определен там, где я и хотел!!! Красота!). Если результаты представить в виде обозначений таксонов, то рис.8. совпадет с тем, что представлено на рис.6:
Рис.8."Таксономическое описание для филогенетического дерева".
Таким образом, при испольвании метода объединения соседей можно доверять укорененному дереву и строить предположения о механизме эволюции. Рис.8Б четко доказывает мое мпредположение, описаное выше для дополненного филогенетического дерева (Рис.6). Если же сравниванать это укорененное дерево (Рис.8Б) с построенным по алгоритму UPGMA (Рис.4Б), то можно заметить, что топология не совпадает и положение корня также отлично. Но неукоренные деревья (сответственно Рис.8А и Рис.4А) находят отличное совпадение. Видимо, такой результат является прямым следствием основного различия в алгоритмах, реализуемых этими программами: UPGMA исследует на минимальность эволюционное расстояние между объектами, а метод Nj: минимальность общей длины ветвей для дерева (при этом пара объектов - ближайшие соседи). Таким образом, напрашивается вывод: если наша цель - описать топологию ветвей и листьев, без укоренения и мы уверены в справедливости "теории молекулярных часов" (видимо, теория справедлива и в моем случае, так как топология неукорененных деревьев по результами обеих методов совпала) то можно вполне доверять результатам алгоритма UPGMA. Если мы не верим этой теории, то доверяем результатам использования алгоритма Nj, конечно, после расчета бутстрепных значений, и неукорененному дереву".
©Володя Рудько