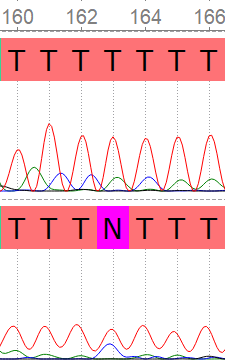
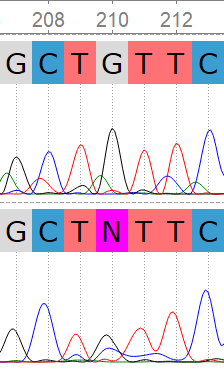
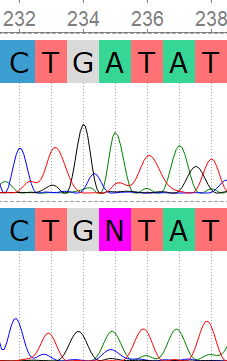
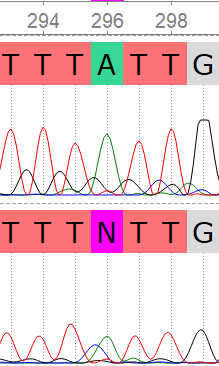
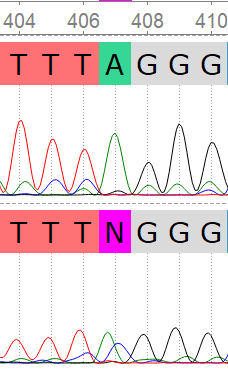
1. Чтение хроматограммы и сборка последовательнсти¶
Исходные файлы: прямой рид, обратный рид
Контиг: консенсусная последовательность в fasta-формате
Выравнивания: неотредактированных ридов с контигом, отредактированных ридов с референсом
В качестве референсной была взята последовательность Macoma balthica voucher 07PROBE-05510 cytochrome oxidase subunit 1 (COI) gene (GeneBank: KF643760.1) полученная с помощью blastn и консенсусной последовательности.