Введение в анализ данных NGS
Описание файла с референсом
Референсный файл - chr3.fna представляет собой стандартный fasta-файл. Первая строчка содержит идентификатор и некоторую
информацию о последовательности. В предложенном файле представлена человеческая хромосома, и первая строчка выглядит так:
>NC_000003.12 Homo sapiens chromosome 3, GRCh38.p13 Primary Assembly
После идёт уже сама последовательность - в начале (и, кажется, в конце) есть большое количество неопределенных нуклеотидов и, судя по всему, теломерных повторов (CCCTAA, но были и немного
отличающиеся повторы, например CCCTCA, СССTAC, вероятно, это какие-то мутации).
Индексирование референса
BWA - пакет инструментов для выравнивания коротких последовательностей с большими референсными геномами, например с человеческим (информацию взял из мануала). Вообще в этом пакете три
алгоритма (BWA-SW, BWA-MEM, BWA-backtrack), но сейчас нам нужен BWA-SW. В этом пункте мы производим индексирование референса. Команда для запуска этого процесса выглядит следующим образом:
bwa index -a bwtsw chr3.fna
bwa index - индексирующая программа, опция -a позволяет выбрать алгоритм (в нашем случае - bwtsw, выбираем, потому что он может работать с большими последовательностями,
такими как человеческий геном). В конце указываем название референсного файла, который мы собираемся индексировать.
На выходе мы получили 5 файлов - chr3.fna.sa, chr3.fna.amb, chr3.fna.ann, chr3.fna.bwt, chr3.fna.pac. В файле chr3.fna.ann находится краткая информация о референсной последовательности,
в файле chr3.fna.amb содержатся нуклеотиды, не являющиеся A, T, G или С. Остальные 3 файла являются бинарными, их посмотреть в текстовом редакторе не удалось, вероятно, они нужны
для последующего картирования.
Описание образца
Был произведён поиск образца в базе NCBI в разделе SRA. Краткую информацию о нём можно увидеть ниже в таблице.
| Пункт | Информация об образце |
| Ссылка на информацию об образце | вот она |
| Прибор | Illumina Genome Analyzer IIx |
| Организм | Homo sapiens |
| Стратегия секвенирования | whole-exome sequencing (в самой строчке Strategy было указано OTHER, но в пункте construction protocol было указано экзомное секвенирование). |
| Чтения | парноконцевые |
| Сколько чтений ожидается? | 41,277,367 |
Проверка качества исходных чтений
Для проверки качества чтений мы воспользовались программой fastqc. Анализировали отдельно каждый файл из двух.
Команды для запуска анализа:
fastqc SRR10720419_1.fastq.gz (ссылка на отчёт)
fastqc SRR10720419_2.fastq.gz (ссылка на отчёт)
Количество пар чтений - 41277367, это число одинаковое у прямых и обратных чтений и совпадает с ожидаемым числом, указанным на сайте NCBI.
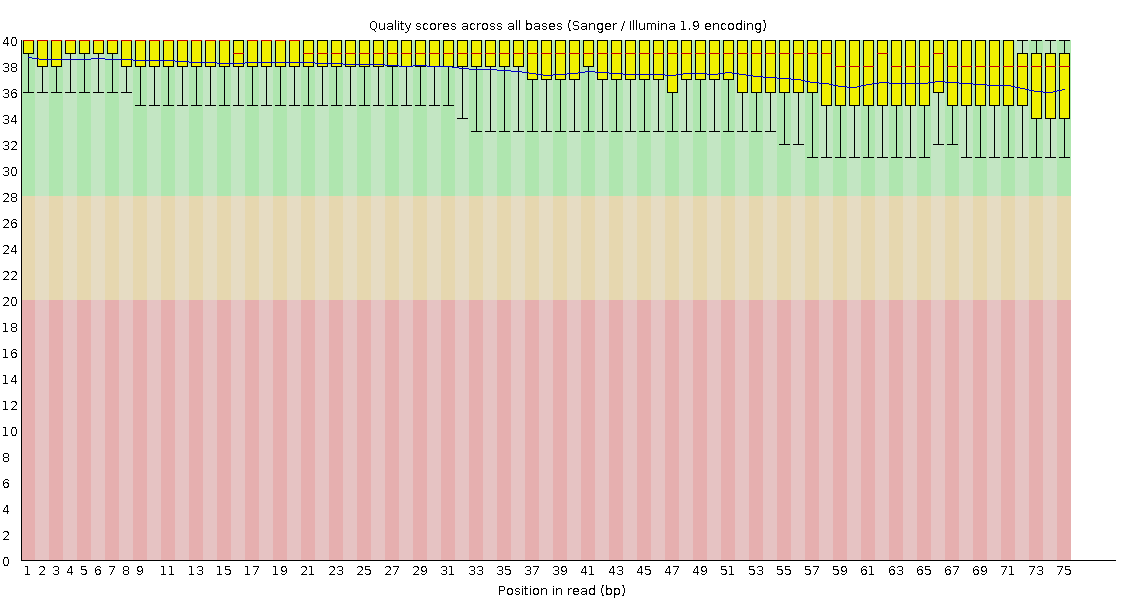
На Рисунках 1 и 2 представлены Графики Per base sequence quality для прямых и обратных чтений соответственно.
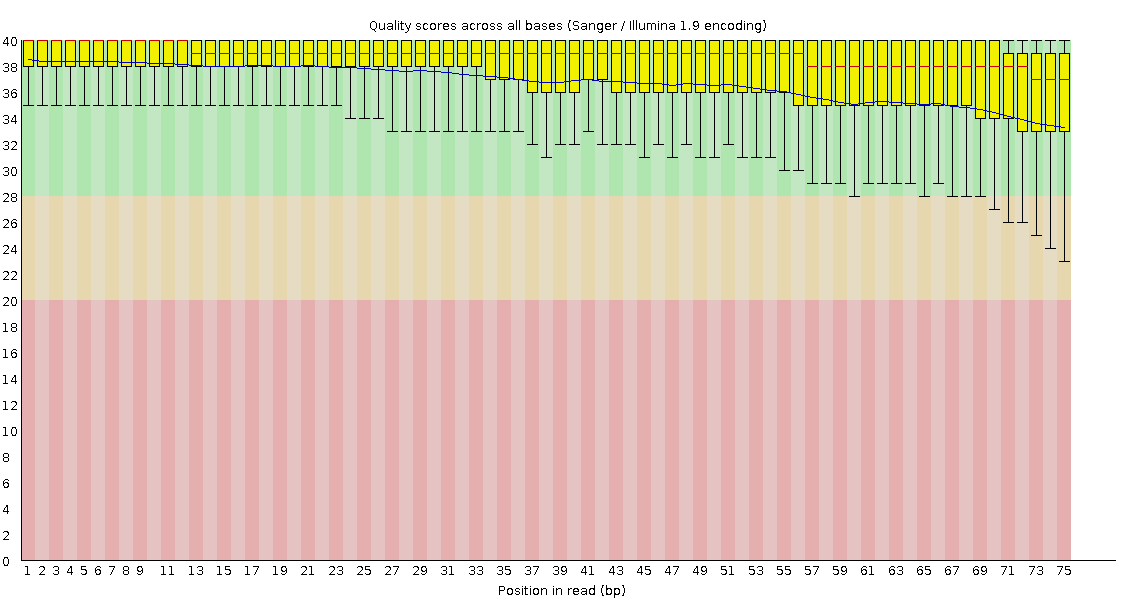
На графиках видно, что значения Phred Quality Score у прямых и обратных чтений лежат в диапазоне от 24 до 40, что нас вполне устраивает, т.к. значение 20 уже даёт нам 99-процентную вероятность
того, что нуклеотид определен верно (информацию взял из презентации).
Ниже представлены графики Per sequence quality score для прямых и обратных чтений. (Рис.5 и 6)
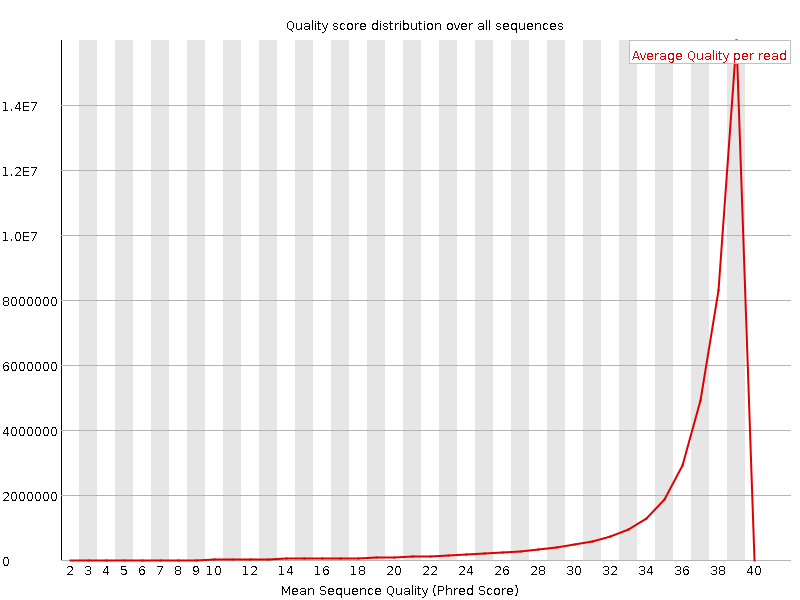
На вышепредставленных графиках видно, что в среднем прочтения имеют Phred-score, равный 39, что говорит о высоком качестве полученных даных. При движении по оси Phred-score влево, количество
прочтений с таким Phred-score резко уменьшается, так, например, чтений с Phred-score меньше 20 нет вообще или совсем мало. В общем, хорошие данные получились.
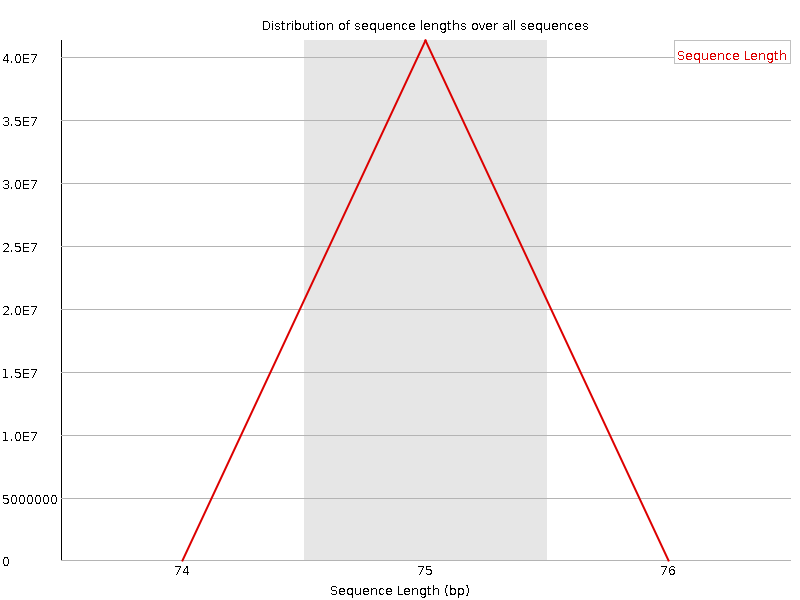
На рисунках 7 и 8 представлены графики Sequence Length Distribution для чтений - на самом деле график для прямых и обратных чтений абсолютно одинаков, но я вставил две
картинки, чтобы это было видно. Судя по графику, подавляющее большинство чтений имеют длину 75 нуклеотидов, что хорошо.
Фильтрация чтений
После анализа чтений мы профильтровали их с помощью программы trimmomatic. Команда для запуска "фильтрации" приведена ниже (выглядит она страшно, честно говоря).
java -jar /usr/share/java/trimmomatic.jar PE -phred33 -threads 10 SRR10720419_1.fastq.gz SRR10720419_2.fastq.gz paired_1.fastq.gz unpaired_1.fastq.gz paired_2.fastq.gz unpaired_2.fastq.gz TRAILING:20 MINLEN:50
Ну и ещё там были введены названия двух входных и четырёх выходных файлов.
Проверка качества триммированных чтений
Для анализа 4 выходных файлов использовалась следующая команда
fastqc unpaired_2.fastq.gz && fastqc unpaired_1.fastq.gz && fastqc paired_1.fastq.gz && fastqc paired_2.fastq.gz
Ссылки на отчёты в формате html:
paired_1.fastqc.html
paired_2.fastqc.html
unpaired_1.fastqc.html
unpaired_2.fastqc.html
В триммированных файлах осталось 39759554 чтения, что составляет 96.32% от исходного количества.
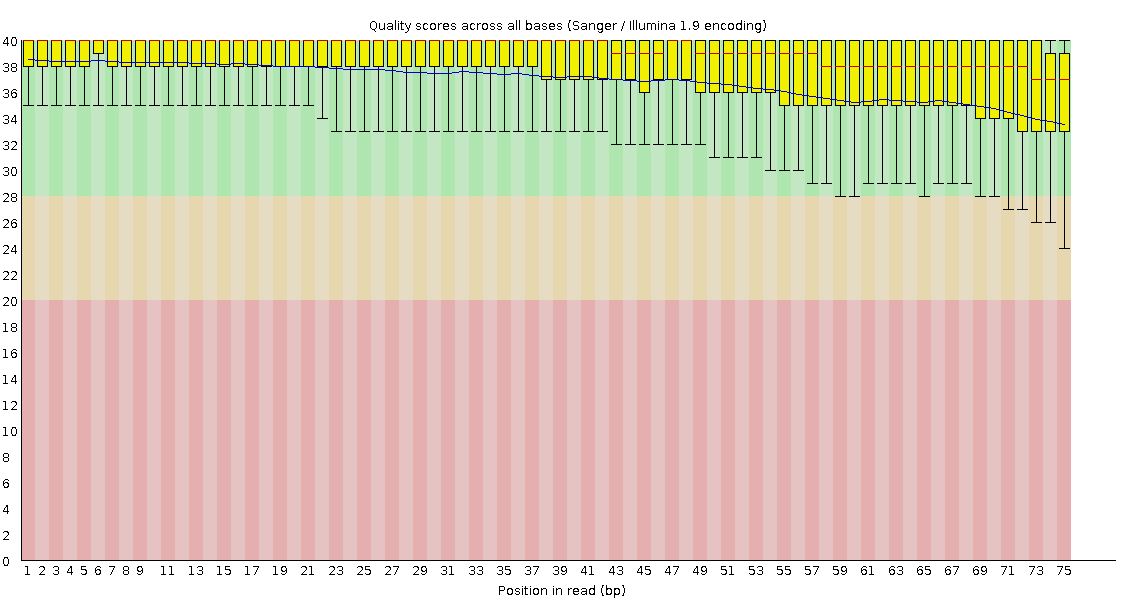
На рисунках 7-10 представлены графики Per Base sequence quality для всех 4 файлов.
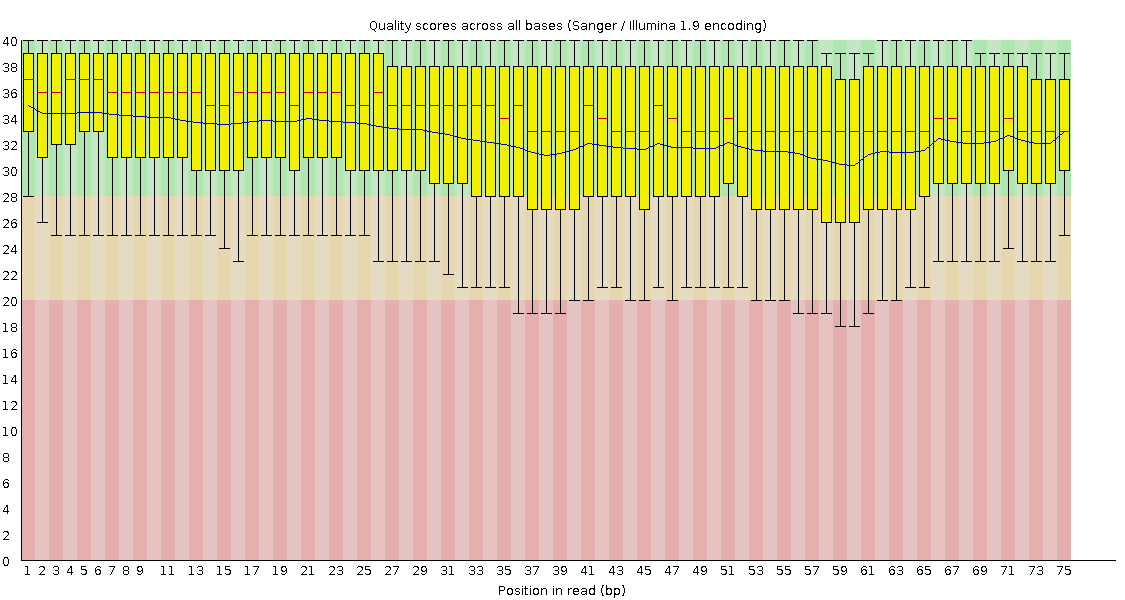
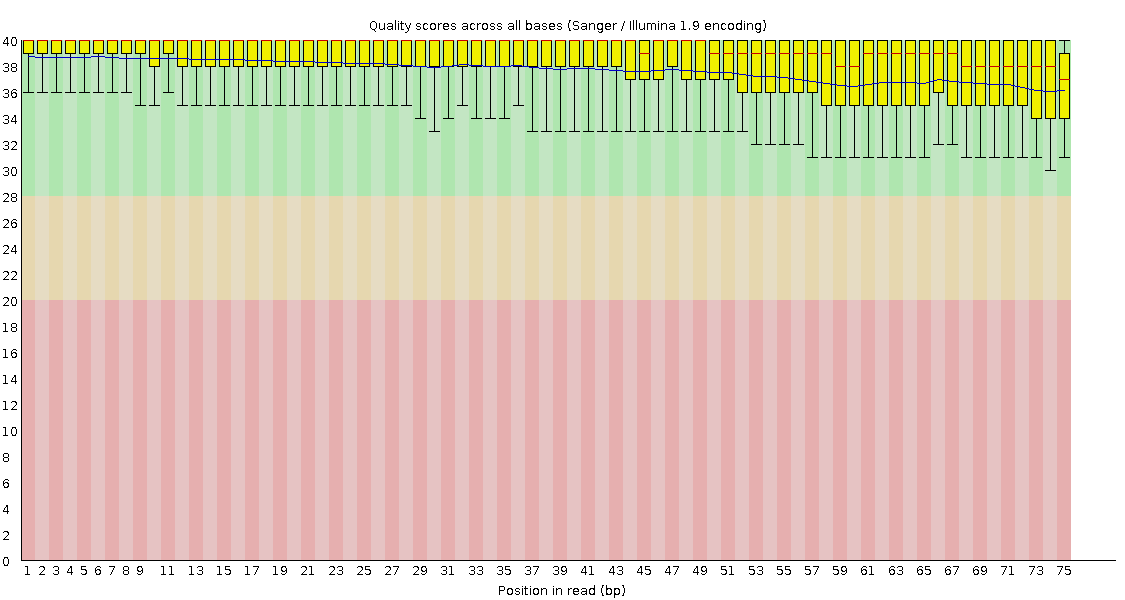
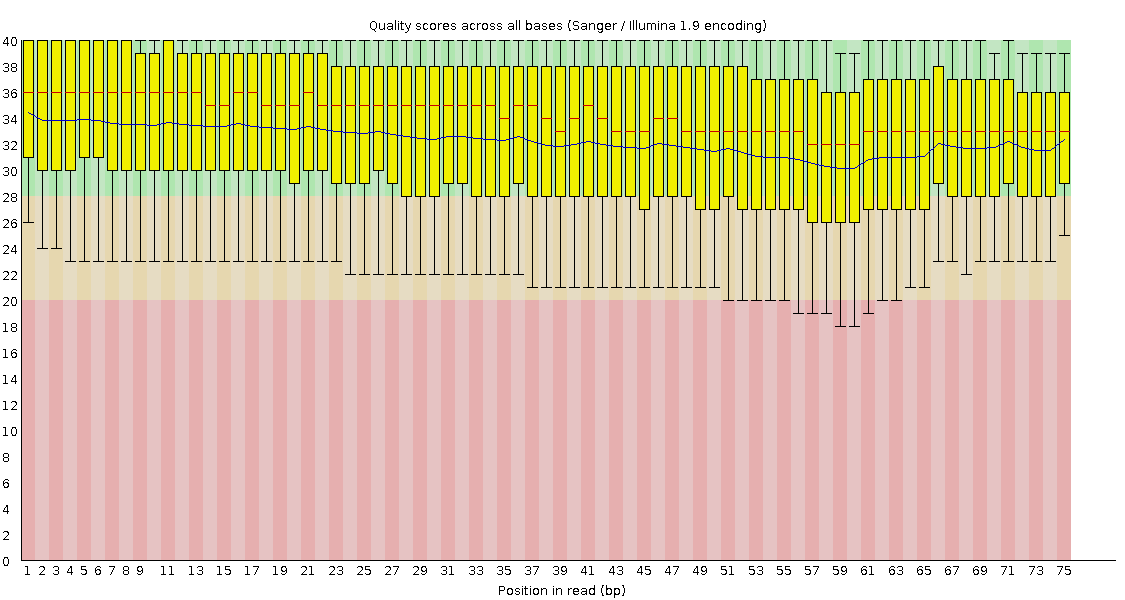
Судя по графикам, качество unpaired чтений в среднем ниже качества paired чтений, как для прямых, так и для обратных чтений. Paired же чтения обладают более высоким качество по сравнению
с нефильтрованными чтениями, особенно сильно это заметно по диапазонам значений. Так, например, у forward paired чтений разброс от 32 до 40, тогда как изначально разброс значений был
от 24 до 40.
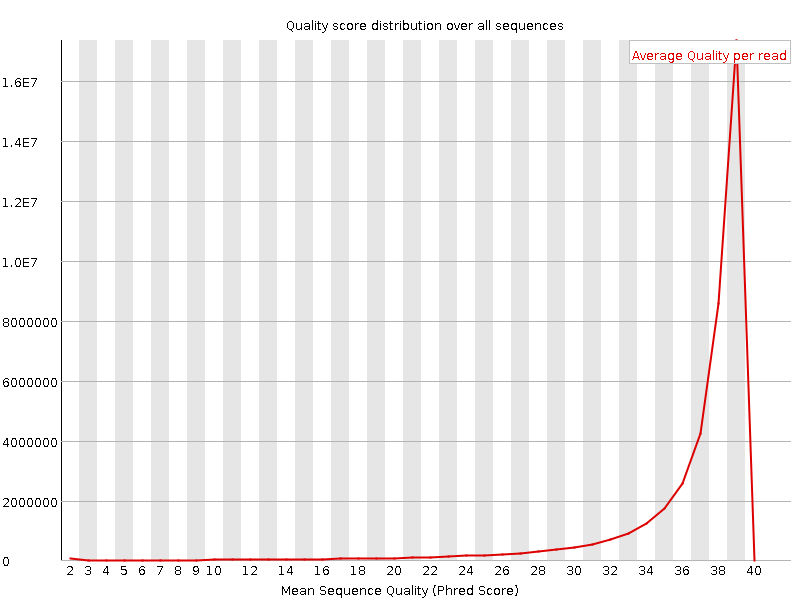
Ниже представлены графики Per sequence quality score (только для paired-чтений)

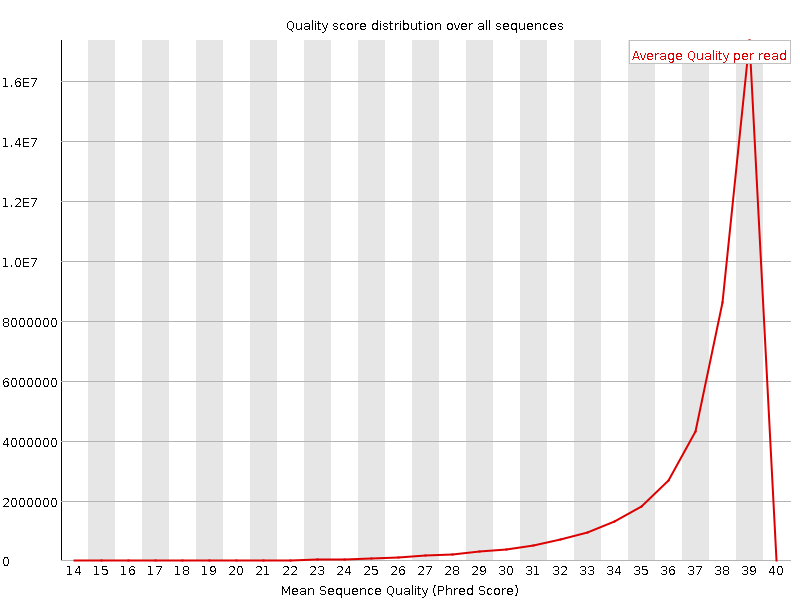
Можно сказать, что график стал более "скученным", так как на для триммированных чтений график начинается на значениях Phred-Score 14 и 18 (на нефильтрованых чтениях график начинался уже
на значениях 2 и 4). Это говорит об уменьшении чтений с низким качеством.
Ещё ниже можно увидеть графики из раздела Sequence Length Distribution
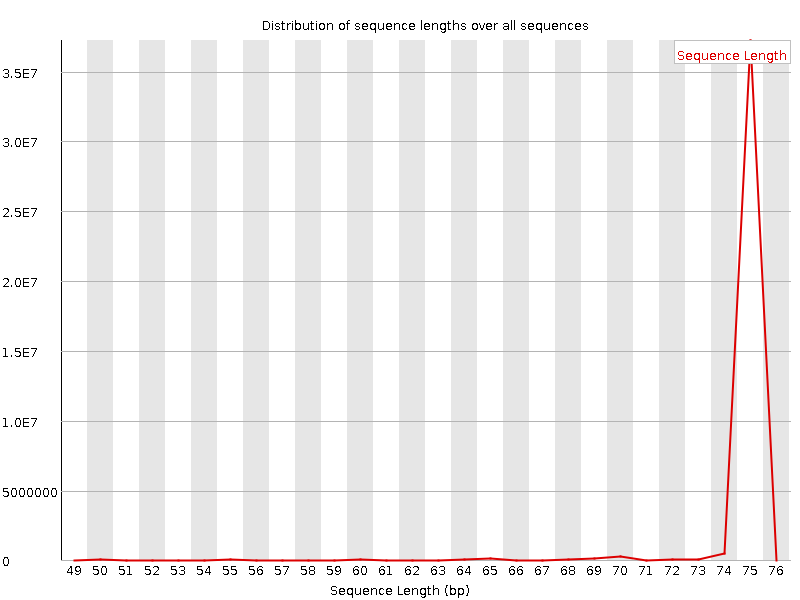
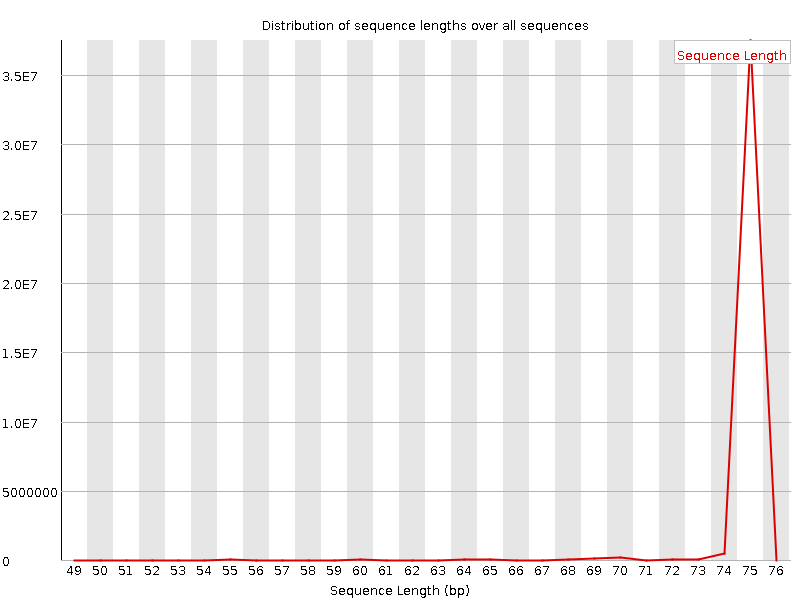
Видно, что распределение длин чтений немного изменилось, график сильно расширился, теперь встречаются чтения с длиной сильно меньше 75, которых не было видно на графике для нетриммированных чтений. Вероятно, это произошло из-за того, что укороченные в результате удаления с конца нуклеотидов с качеством меньше 20 чтения приобрели больший Phred-Score.