Введение в транскриптомный анализ
Подготовка референса.
Для подготовки референса была введена команда:
hisat2-build chr3.fna chr3 2> log_hisat_index.txt
На вход была подана fasta-файл с референсом, на выходе получены 8 бинарных файлов индексирования и файл log_hisat_index.txt, куда мы направили поток ошибок.
Оценка качества чтений.
Для получения html-отчёта о качестве чтений была использована команда:
fastqc SRR2015718_1.fastq.gz
Файл с html-отчётом можно посмотреть
здесь.
Всего в файле 33 065 729 чтений. Ниже, на Рисунке 1, представлен график Per base sequence quality для чтений.
Видно, что большинство боксплотов лежат в зелёной области, что говорит о хорошем качестве чтений (с ними можно
работать).

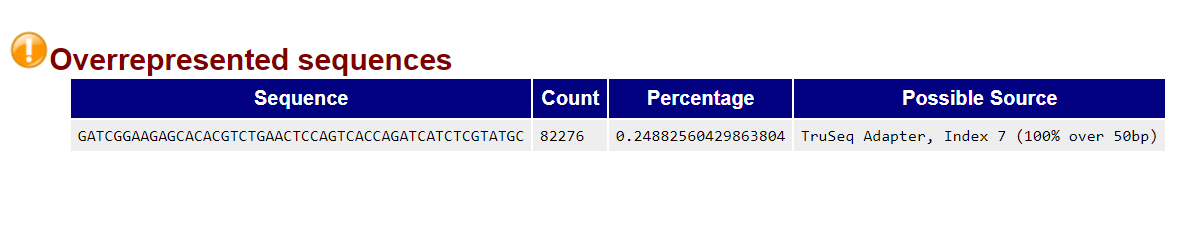
Что меня встревожило, так это графа Overrepresented sequences, в которой говорится о слишком большом количестве последовательности адаптера в чтениях. Наверное, это плохо.
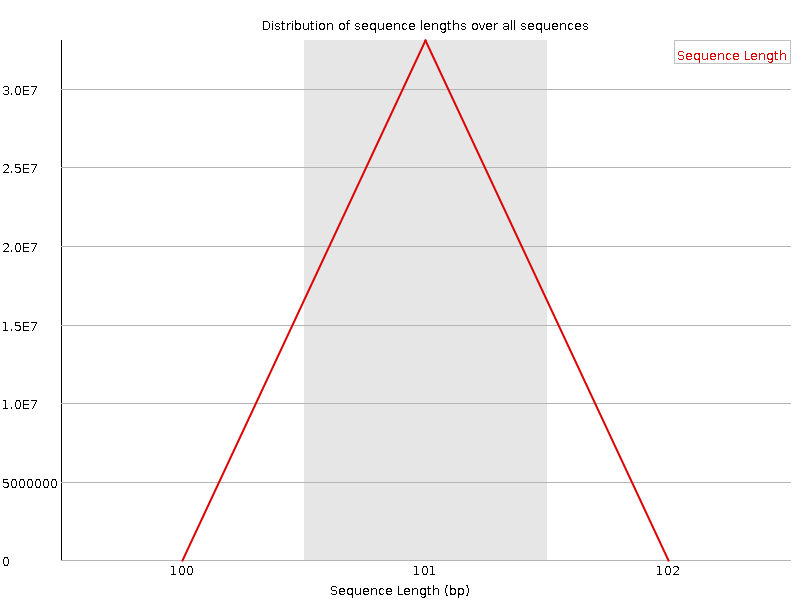
На Рис.3 представлено распределение чтений по длине - здесь всё хорошо и приятно, подавляющее число ридов (больше 30 миллионов) имет длину в 101 нуклеотид.
Картирование чтений на референс.
Для картирования чтений на референс была выполнена следующая команда:
hisat2 -p 10 -x chr3 -k 3 -U SRR2015718_1.fastq.gz > rna_mapped.sam 2>log_er.txt
Файл log_er.txt можно посмотреть здесь. В этой команде:
Согласно log-файлу, 31201546 (94.36%) чтений не было картировано, что логично, т.к. мы картируем тотальную РНК только на одну
хромосому. К тому же, РНК может отличаться, и достаточно сильно, от гена на хромосоме из-за сплайсинга. На референс было картировано 1843520 (5.58%) чтений.
После этого sam файл был переведён в bam формат и отсортирован с помощью следующих команд:
samtools sort -o rna_mapped.bam rna_mapped.sam
samtools index -@ 10 rna_mapped.bam
Затем были отобраны только чтения, которые легли на 3-ю хромосому. Это было сделано с помощью команд:
samtools view -@ 10 -h rna_mapped.bam NC_000003.12 > rna_chr3.sam
samtools view -@ 10 -b rna_chr3.sam > rna_chr3.bam
Поиск экспрессирующихся генов.
Для поиска генов мы скопировали файл gencode.chr3.gtf, в котором есть разметка генов для 3-ей хромосомы.
Этот файл состоит из шапки с информацией о базе данных, формате файла, дате публикации данног файла, и таблицы со следующими колонками:
Для каждого гена из разметки было посчитано количество картированных на него чтений. Сделано это было с помощью команды:
htseq-count -f bam -s no -t exon rna_chr3.bam gencode.chr3.gtf 1> htseq
В этой команде:
После этого с помощью команды wc -l htseq | head -n $linea-5 htseq | awk '{s+=$2}END{print s}' было посчитано, что в границы генов попало 1466440 чтений.
Командой tail htseq мы просмотрели конец файла htseq, чтобы найти данные о количестве ридов, не попавших в границы генов - таких ридов оказалось 312885.