Сборка генома de novo
Получение чтений и общего файла с адаптерами
Для получения файла с ридами была использована следующая команда:
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/001/SRR4240381/SRR4240381.fastq.gz
После этого последовательности адаптеров были скопированы из папки adapters в рабочую папку и объединены в общий файл. Для этого использовались следующие команды:
cp /mnt/scratch/NGS/adapters/* /mnt/scratch/NGS/yablinkubovich/de_novo
cat *.fa » all_adap.fasta
Очистка ридов с помощью trimmomatic
В следующей части работы мы проводили очистку ридов с помощью программы trimmomatic. Для начала мы удалили из ридов адаптерные последовательности с помощью следующей команды:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 10 SRR4240361.fastq.gz cleaned1.fastq.gz ILLUMINACLIP:all_adap.fasta:2:7:7 2> log.txt
Файл с логом можно посмотреть здесь. Видно, что процедура прошла успешно (что радует), и
было удалено 34532 чтения, что составляет 0.47% от общего количества (осталось, соответственно, 99,53% чтений).
На этом мы не остановились, и следующим шагом было удаление с правого конца чтений нуклеотидов с качеством меньше 20, и последующее удаление чтений с длиной меньше 32 нуклеотидов. Команда,
выполняюшая эту фильтрацию, представлена ниже:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 -threads 10 cleaned.fastq.gz filtered.fastq.gz TRAILING:20 MINLEN:32 2> log2.txt
Файл с логом можно посмотреть здесь. Видно, что в результате фильтрации осталось 6834335 (94.42%)
чтений. Итого, всего было удалено 438 286 чтений (приблизительно 6 процентов от изначального количества).
Размер файла:
Запуск velveth
Для создания k-меров на основе наших чтений длиной 31 нуклеотид была введена следующая команда:
velveth velveth 31 -fastq -short filtered.fastq.gz
В этой команде -fastq задаёт формат входного файла, -short говорит о том, что короткие чтения. На выходе получили директорию velveth с несколькими файлами в ней.
Запуск velvetg
Была запущена программа velvetg. Команда запуска представлена ниже:
velvetg velveth
Лог можно посмотреть здесь. Из него можно получить информацию о N50 - значение этого параметра
равно 25683. Также в результате выполнения этой программы появились файлы contigs.fa и stats.txt. В файле contigs.fa представлены сами контиги, в stats.txt - статистика
по этим контигам.
Среднее покрытие равно 481.945.
Информация о самых больших контигах была получена с помощью команды:
sort -n -r -k 2 stats.txt | head
Ниже представлена информация о трёх самых длинных контигах.
| ID контига | Длина | Покрытие |
| 6 | 49238 | 26.66 |
| 2 | 45555 | 26,45 |
| 34 | 43866 | 23,51 |
Контиг с ID 62 имеет аномально большое покрытие, равное 212829, однако это объясняется его длиной, равной единице (странно). Контиг с самым маленьким покрытием имеет длину 4 нуклеотида и покрытие, равное 1.5.
Megablast для трёх самых длинных контигов
C помощью утилиты seqretsplit из файла contigs.fa были получены интересующие нас контиги (их нуклеотидные последовательности), а именно контиги с ID 2,6 и 34. После этого было произведено сравнение программой megablast (так как мы имеем дело с очень схожими последовательностями) с референсным геномом. В Таблице 2 можно найти информацию о трёх произведённых выравниваниях.
| ID контига | E-value | Query Cover | Identity | Total Score | Ссылка на отчёт |
| 6 | 0.0 | 80% | 74.95% | 21380 | файл |
| 2 | 0.0 | 70% | 77.02% | 17272 | файл |
| 34 | 0.0 | 71% | 78.79% | 16707 | файл |
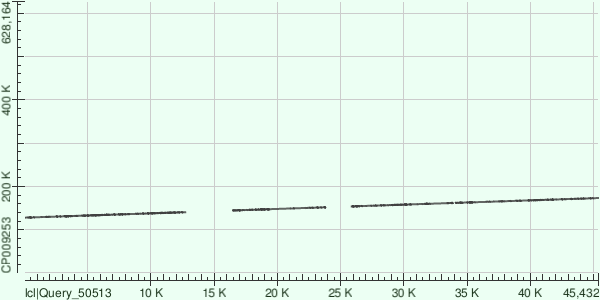
Контиг 6 соответствует референсному геному на участке с 127825 по 173180 нуклеотид. В лучшем выравнивании - 548 гэпов.
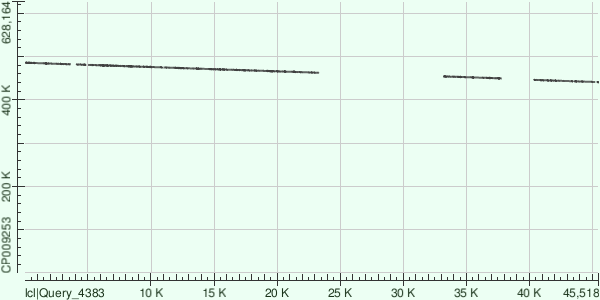
Контиг 2 соответствует референсному геному на участке с 266073 по 285070 нуклеотид. В лучшем выравнивании 208 гэпов. В этом выравнивании интерес представляет то, что контиг выровнялся в "инвертированном" состоянии, однако у нас в данных нет информации о цепи, поэтому мы не можем сказать, что на этом участке произошла какая-либо инверсия.
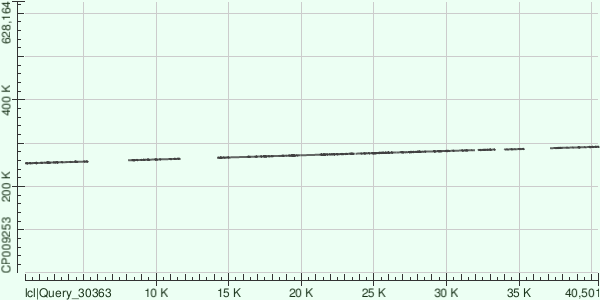
Контиг 34 соответствует референсному геному на участке с 266073 по 285070 нуклеотид. В лучшем выравнивании 361 гэп.