Секвенирование по Сэнгеру
В этой работе необходимо было проанализировать результаты секвенирования по Сэнгеру и на основании данных о прямой и обратной цепочках ДНК создать консенсусную последовательность.
Сначала оба выданных файла с прямой и обратной цепочками были загнаны в Pearl, чтобы посмотреть, что это вообще даст. И дало это много - почти выполненное задание (как впоследствии оказалось, не совсем почти).

Позже оказалось, что у других программ, в частности, у Chromas, есть свои преимущества - в Chromas выделяются нечитаемые участки 5' и 3' концов, и ещё там можно двигать хроматограммы независимо друг от друга. После Pearl хроматограммы были загружены в Chromas и оттуда сохранены в формате fasta (при таком сохранении автоматически убираются нечитаемые концы). Таблица с границами нечитаемых участков представлена ниже.
| Участок в начале | Участок в конце | |
| Прямая цепочка | 1-18 нуклеотид (18bp) | 376-379 нуклеотид (4 bp) |
| Обратная цепочка | 1-32 нуклеотид (32 bp) | там все нормально читается, проблем нет |
По поводу автоматического определения нечитаемых участков (они определялись в Chromas) можно сказать, что они совпадают с теми участками, которые можно определить на глаз.
В начале прямой цепочки на границе нечитаемого участка (он выделен слабым оранжевым) виден переход от полностью нечитаемой хроматограммы к более определенной, в которой
уже можно определить нуклеотид.
Так же и в остальных - границы, определенные на глаз, отличаются от программных не более, чем на 1-2 нуклеотида.


Затем обратная цепочка была инвертирована с заменой нуклеотидов на комплементарные в программе JalView. После этого две цепочки были выровнены с помощью программы
muscle (fasta-файл) и визуализированы в Jalview.

В целом, хроматограмма хорошая: шум небольшой, появляется только в начале прямой цепи, почти везде можно однозначно определить нуклеотид.
Теперь необходимо было посмотреть на проблемные места (в частности, несовпадения в выравнивании) и проанализировать их вручную. Для этого в Chromas
параллельно были открыты обе
цепочки и двигались друг с другом. Важно учитывать, что прямая цепочка на всех фотографиях расположена сверху (обратная, что логично, снизу).
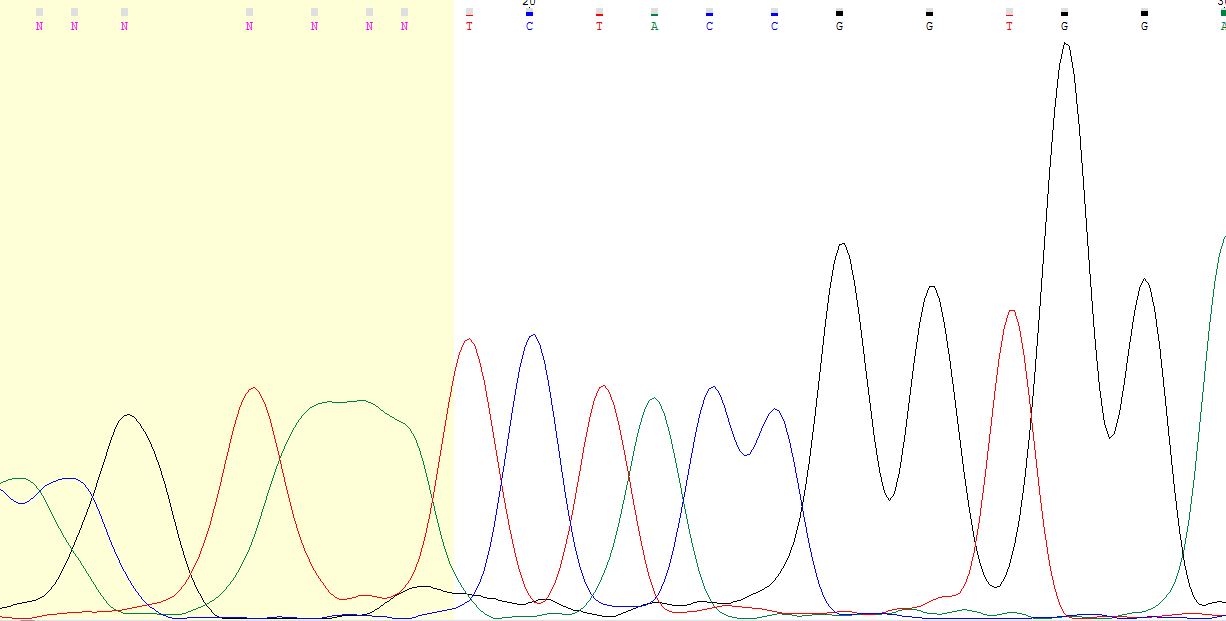
Первый проблемный участок находится в начале последовательности (12-14 на прямой цепочке, 61-63 на обратной)
- как видно на фото, в начале с хроматограммой у прямой цепочки не всё хорошо (хотя Chromas её удалять
не стал), зато всё чётко видно на обратной цепочке (там аденины).
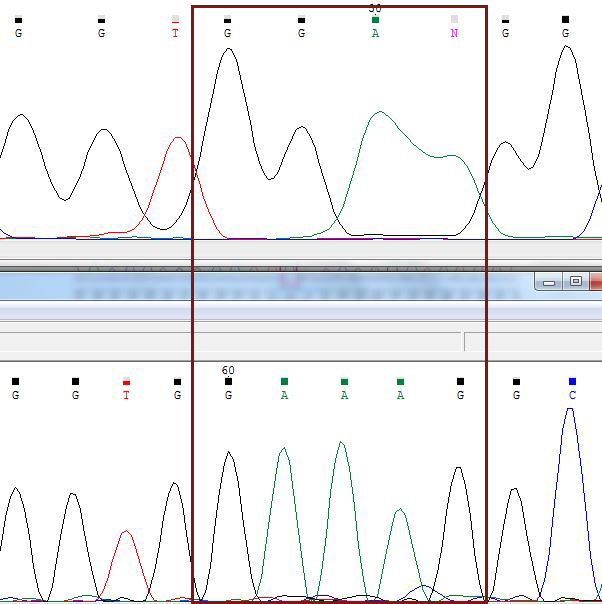
Как мне кажется, полиморфизм наблюдается на 50 нуклеотиде на прямой цепочке - 100 нуклеотиде на обратной (в дальнейшем буду как-то эту запись сокращать). Обе цепочки показывают двойной пик А и С, поэтому нельзя точно сказать, какой именно нуклеотид стоит в этой позиции, вероятно, это правда полиморфизм (М).
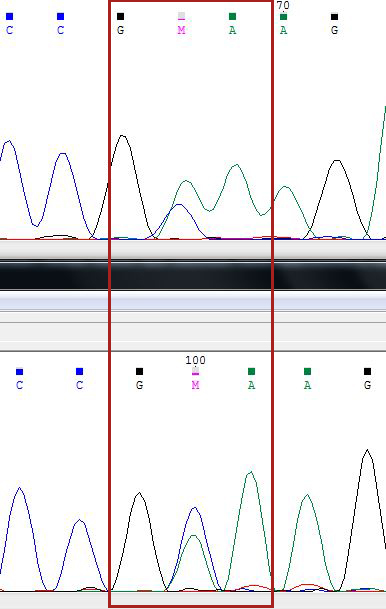
59 п.ц - 109 о.ц (далее п.ц. - прямая цепочка, о.ц. - обратная цепочка) - на прямой цепи в этот момент происходит что-то непонятное (может быть, из-за цитозинов вокруг искомого нуклеотида сигнал от них ещё не исчез во время детекции), на обратной цепочке ясно виден гуанин. Его и вставим в консенсусную последовательность.

70 п.ц. - 120 о.ц. - полиморфизм, вероятнее всего, т.к. на обеих цепях есть два одновременнх пика А и Т (отмечаем по номенклатуре как W).

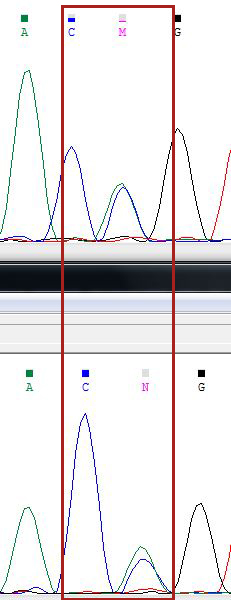
107 п.ц. - 157 о.ц. - полиморфизм, два одновременных пика А и С (отмечаем по номенклатуре как M).Странно, что на обратной цепочке это место отмечено как N, т.к. там одинаковые ситуации. Большего про это место сказать нельзя.
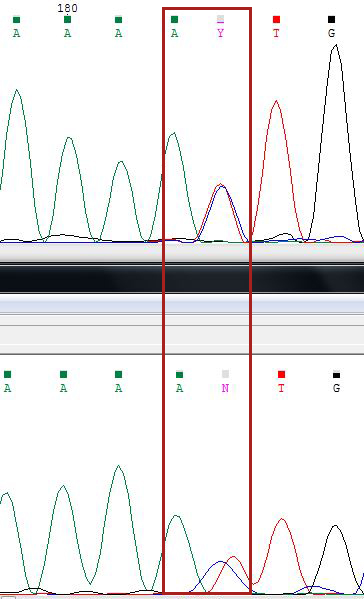
165 п.ц. - 215 о.ц. - полиморфизм, два одновременных пика T и С (отмечаем по номенклатуре как Y).
После всех исправлений была получена консенсусная последовательность,которую
можно скачать по этой ссылке.
Выравнивание почищенной прямой и обратной цепочек можно скачать здесь
Задание 2
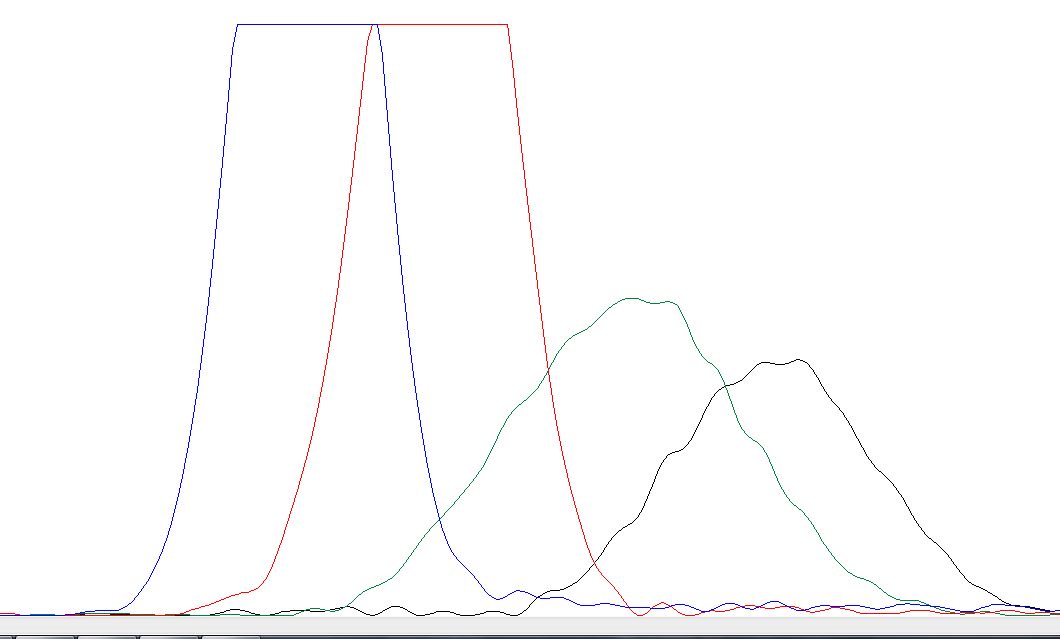
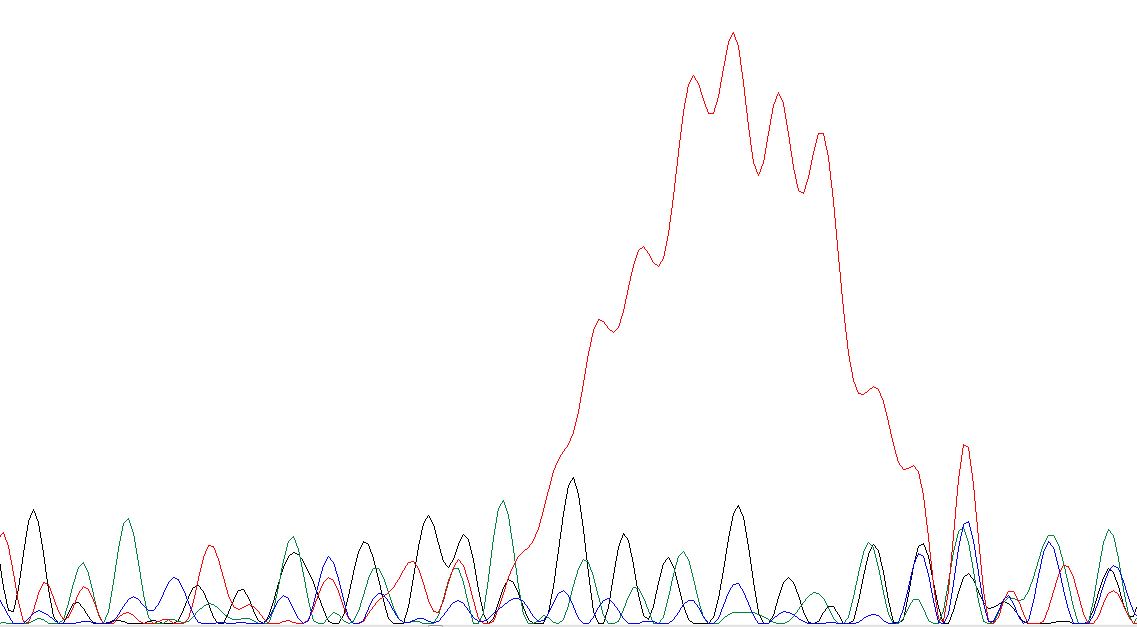
Для ознакомления с примерами очень плохих хроматограмм я зашёл в директорию bad и взял оттуда вот
этот файл. В нём и правда всё очень плохо, настолько плохо, что Chromas даже не
может определить, где ставить нуклеотиды (на картинках видно, что программа даже не пишет N, так как просто не понимает, куда). Видно, что попало несколько образцов ДНК
в пробу, ещё видны какие-то аномально высокие и протяженные пики (может быть, неисправность прибора, или пятно краски). Ниже представлены самые забавные участки,
найденные мной.