Алгоритмы и программы множественного выравнивания.
Для анализа было выбрано семейство Цистатинов (Cystatin, Pfam: PF00031). Выравнивание строилось для трех белков с известной 3D-структурой:
- 1a67 — Цистатин куриного яичного белка (птицы)
- 1g96 — Цистатин C человека (млекопитающие)
- 2kxg — Цистатин из семян тыквы (растения)
Сравнение проводилось для трех программ: A (MUSCLE), B (MAFFT) и C (T-Coffee) с помощью программы для сравнения выравниваний.
Результаты сравнения выравниваний
1. Сравнение выравниваний A (MUSCLE) и B (MAFFT)
- Длина выравнивания A (MUSCLE): 121 колонка
- Длина выравнивания B (MAFFT): 128 колонок
- Количество совпадающих колонок: 99
- Процент совпадения: 81.8% (от длины A) | 77.3% (от длины B)
Cписок блоков одинаково выровненных колонок (длина ≥ 2):
- (10,60) = (10,60) — длина: 51
- (75,121) = (82,128) — длина: 47
Список одинаково выровненных колонок, не входящих в блоки (одиночные совпадения):
(1,1)
Несовпадающие участки (в координатах MUSCLE):
- 1-9 (длина 9)
- 61-74 (длина 14)
2. Сравнение выравниваний A (MUSCLE) и C (T-Coffee)
- Длина выравнивания A (MUSCLE): 121 колонка
- Длина выравнивания C (T-Coffee): 121 колонка
- Количество совпадающих колонок: 94
- Процент совпадения: 77.7% (от длины A) | 77.7% (от длины C)
Cписок блоков одинаково выровненных колонок (длина ≥ 2):
- (81,121) = (81,121) — длина: 41
- (7,36) = (7,36) — длина: 30
- (53,63) = (53,63) — длина: 11
- (69,78) = (69,78) — длина: 10
Список одинаково выровненных колонок, не входящих в блоки (одиночные совпадения):
(2,2), (4,4)
Несовпадающие участки (в координатах MUSCLE):
- 1-6 (длина 6)
- 37-52 (длина 16)
- 64-68 (длина 5)
- 79-80 (длина 2)
Ссылки на материалы:
- Выравнивание A (MUSCLE) в формате FASTA
- Выравнивание B (MAFFT) в формате FASTA
- Выравнивание C (T-Coffee) в формате FASTA
- Проект Jalview со всеми выравниваниями
Таким образом, Выравнивание B (MAFFT) больше похоже на выравнивание A (MUSCLE), чем выравнивание C (T-Coffee). Об этом можно судить по проценту совпадения колонок (81,8% против 77,7%) и по степени фрагментации (сравнение MUSCLE и MAFFT выделило всего 2 огромных непрерывных блока совпадений, в то время как при сравнении с T-Coffee выравнивание разбилось на 4 коротких блока).
Выравнивание по совмещению структур и его сравнение с выравниванием программой MSA
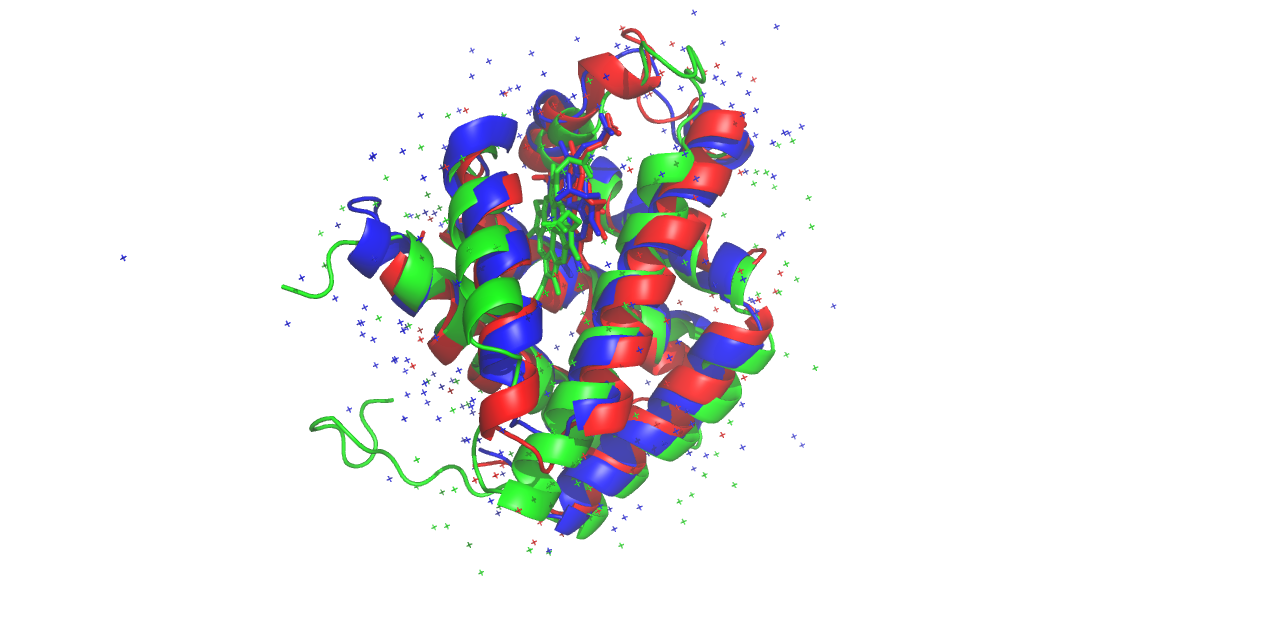
Для анализа было выбрано семейство Глобины (InterPro: IPR000971). Выравнивание строилось для трех белков с:
- 1emy — Миоглобин азиатского слона
- 1ebt — Гемоглобин моллюска
- 1d8u — Гемоглобин риса
Пространственное совмещение структур
Совпадающие блоки колонок выравниваний
На основе сравнительного анализа эталонного 3D-выравнивания (PDBeFold) и текстового выравнивания по последовательностям (Muscle), проведенного с помощью скрипта prakt-D-compare_aln.py, с помощью программы для сравнения выравниваний было обнаружено 6 совпадающих блоков (общее число совпадающих колонок — 113, что составляет 62,1% от длины выравнивания 3D-Aln и 66,1% от длины выравнивания Muscle-MSA).
Cписок блоков одинаково выровненных колонок (длина ≥ 2):
- (13,55) в 3D-Aln = (13,55) в Muscle-MSA — длина: 43
- (157,182) в 3D-Aln = (146,171) в Muscle-MSA— длина: 26
- (126,145) в 3D-Aln = (115,134) в Muscle-MSA— длина: 20
- (111,121) в 3D-Aln = (100,110) в Muscle-MSA— длина: 11
- (1,10) в 3D-Aln = (1,10) в Muscle-MSA— длина: 10
- (71,72) в 3D-Aln = (65,66) в Muscle-MSA— длина: 2
Одиночных совпадений вне блоков:
(148,137)
Несовпадающие участки:
- 11-12 (длина 2)
- 56-70 (длина 15)
- 73-110 (длина 38)
- 122-125 (длина 4)
- 146-156 (длина 11)
Сравнение двух выравниваний показало, что они во многом совпадают, но в некоторых местах сильно различаются. Наличие крупных совпадающих блоков (длиной 43, 26 и 20 колонок) говорит о том, что в этих участках последовательности выбранных белков сохранили высокую гомологию. Текстовый алгоритм Muscle смог правильно сопоставить аминокислоты по их сходству, и этот результат полностью совпал с физическим наложением атомов в пространстве.
Несовпадающие участки (например, регионы длиной 15 и 38 колонок) показывают места, где текстовый алгоритм Muscle совершил ошибки. Так как выбранные глобины принадлежат эволюционно далеким организмам (слон, моллюск, рис), их последовательности на этих участках сильно изменились. Muscle пытался формально подогнать буквы друг под друга и неверно расставил гэпы (прочерки). Структурное выравнивание PDBeFold оказалось точнее, так как оно совмещает белки по реальным 3D-координатам атомов, независимо от схожести букв.
Таким образом, структура белков эволюционно гораздо более консервативна, чем их первичная последовательность.
Ссылки на материалы:
- Выравнивание по совмещению структур в формате FASTA (PDBeFold)
- Выравнивание последовательностей в формате FASTA (Muscle)
- Файл проекта Jalview (.jvp) со сравниваемыми выравниваниями
Краткое описание программы MUSCLE (MUltiple Sequence Comparison by Log-Expectation)
Основные особенности алгоритма:
Алгоритм MUSCLE относится к категории прогрессивных методов множественного выравнивания и включает три основные стадии вычислительного процесса [1]:
- Стадия быстрого приближенного выравнивания: программа проводит оценку эволюционных расстояний между последовательностями на основе подсчета коротких слов (k-mer counting) и строит по ним быстрое направляющее дерево для создания чернового базового выравнивания.
- Стадия улучшения дерева: на основе полученного выравнивания расстояния пересчитываются точнее (метод Kimura distance), дерево перестраивается, и прогрессивное выравнивание повторяется с использованием логарифмической весовой функции (log-expectation score).
- Стадия итеративного уточнения: построенное дерево последовательно разделяется на поддеревья, выделенные профили перевыравниваются между собой. Если новая математическая оценка выравнивания оказывается лучше предыдущей, изменения фиксируются. Процесс повторяется до стабилизации оценки.
Преимущества и эффективность:
Программа обеспечивает высокое качество выравнивания при низких затратах процессорного времени и памяти [1]. Согласно независимому бенчмарку на тестовых наборах BAliBASE, программы MUSCLE и CLUSTALW были признаны самыми быстрыми среди популярных инструментов множественного выравнивания [2]. При этом итеративное уточнение в MUSCLE позволяет исправлять ошибки ранних этапов прогрессивного выравнивания, обеспечивая более высокую точность и надежность результатов на сложных датасетах по сравнению с классическими эвристическими методами [2].
Источники информации:
- [1] Robert C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Research, Volume 32, Issue 5, 1 March 2004, Pages 1792–1797,https://doi.org/10.1093/nar/gkh340
- [2] Pais, F.SM., Ruy, P.d.C., Oliveira, G. et al. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol 9, 4 (2014), https://doi.org/10.1186/1748-7188-9-4