Был выбран домен RVT_N - N-концевой домен
бакткриальных обратных транскриптаз. В скачанном seed было 78 последовательностей, после исключения совпадающих на
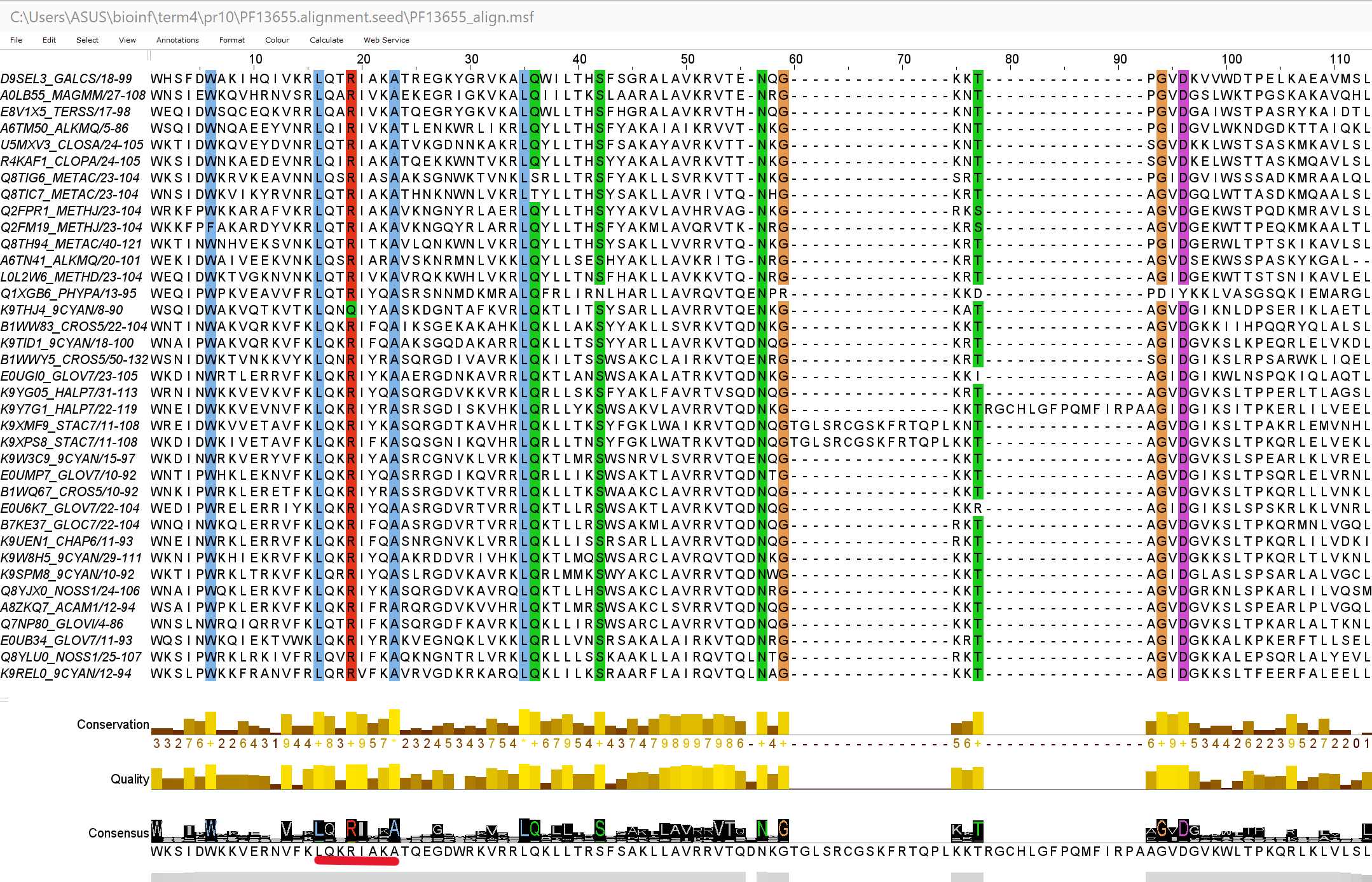
95% ни одна не была убрана. На рис. 1 можно увидеть выравнивание seed и выбранный мной мотив(отмечено красным). Его
паттерн Jalview: [LM][QR].R[IVL][AYFV][KRQAH]A Паттерн Prosite: [LM]-[QR]-x-R-[IVL]-[AYFV]-[KRQAH]-A.
При поиске по этому паттерну находится 71 последовательность.
Рис.1 Выравнивание seed и выбранный мотив(отмечен красным).
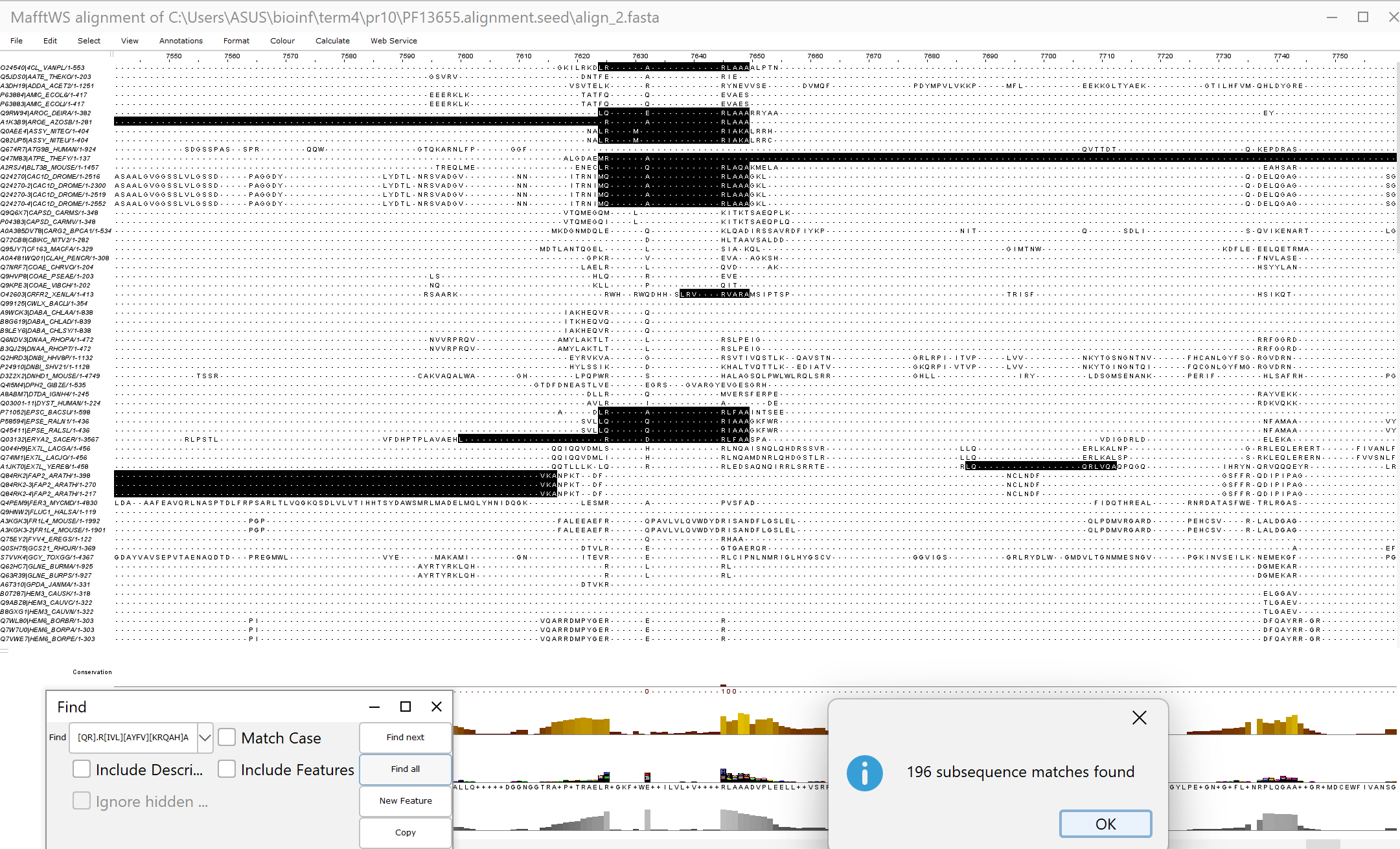
Поиск по паттерну в Prosite дал 198 результатов, два из которых пришлось удалить, потому что Jalview не знает,
что такое селенцистеин. Оставшиеся 196 белков были выровнены программой MAFFT со стандартными параметрами. Как
видно по рис. 2, домены правильно не выровнялись.
Рис.3 Выравнивание MAFFT найденых в Prosite белков. Выделен паттерн, по которому их искали.
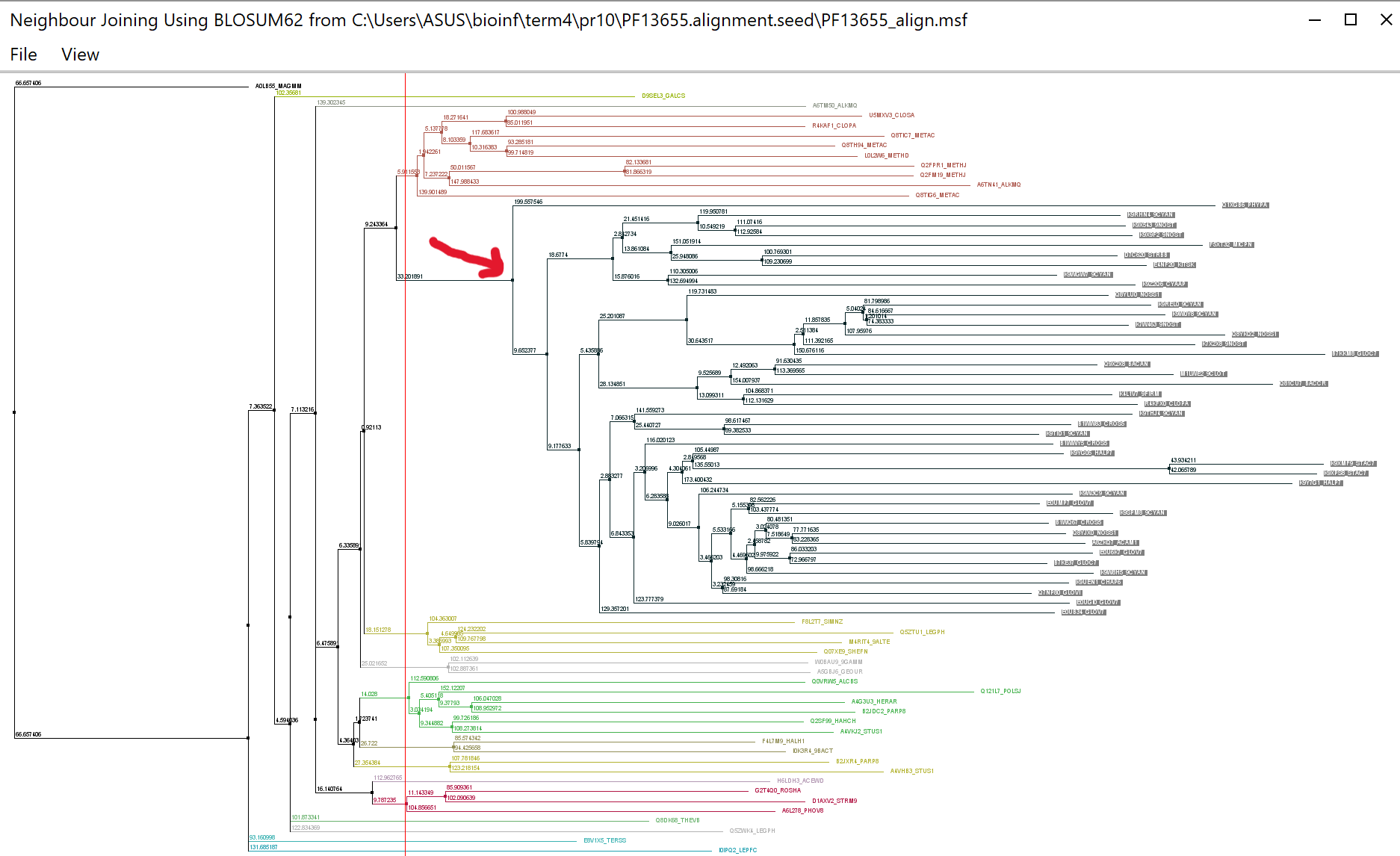
Построим алгоритмом NJ дерево по выравниванию seed (рис. 3). Рассмотрим кроновую группу: она располагается на
весьма длинной ветви, на основании чего можно предположить, что у её представителей присутствует эволюционно
продвинутый мотив.
Рис.4 Дерево seed и.
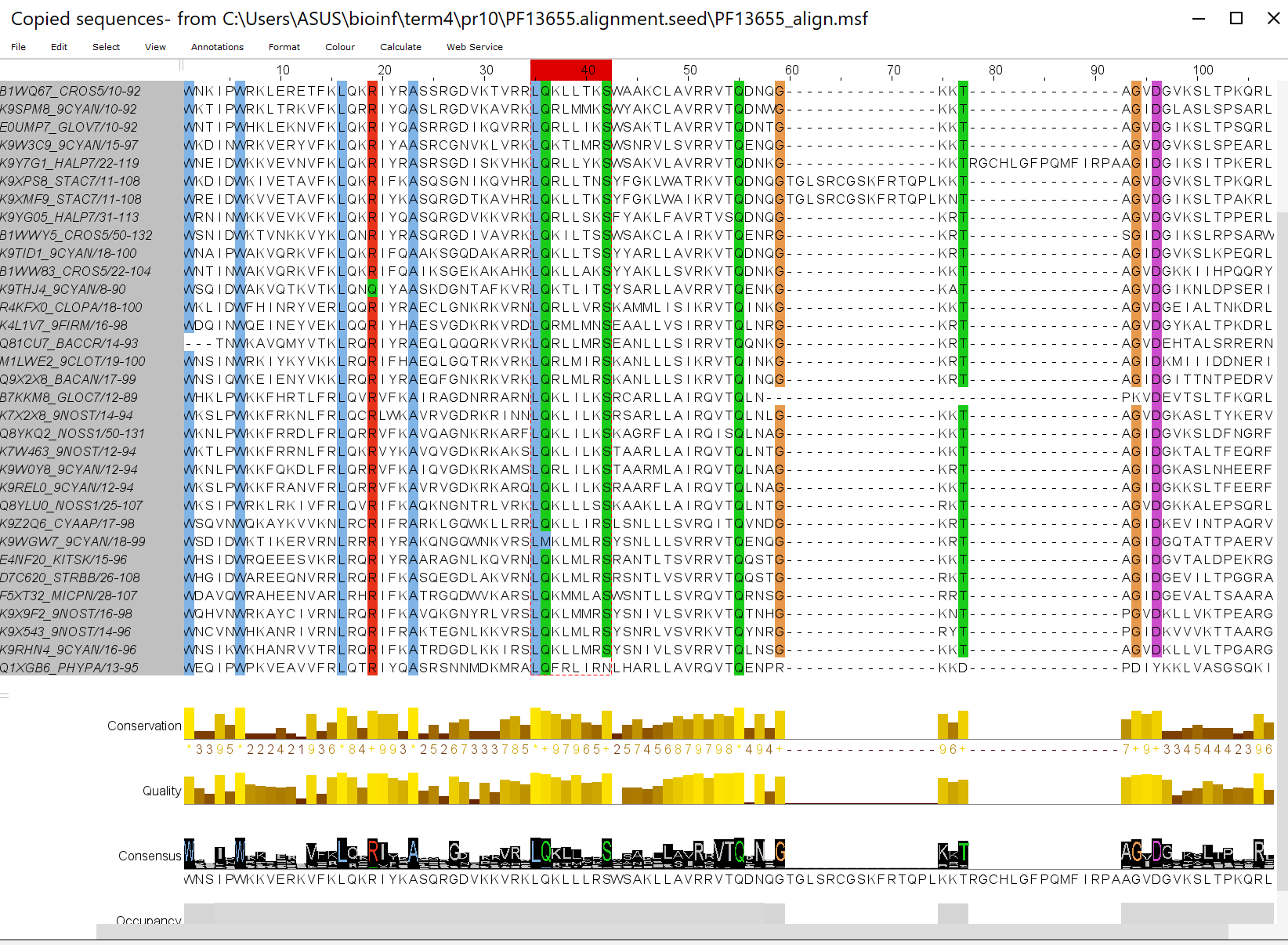
Теперь вынесем представителей кроновой группы из 42 представителей в отдельное выравнивание(рис. 5). Отчётливо виден мотив
L[QM][KRF][LTMIR][LMI][LMITAVSY][RKNSAHQT][SN]. В общем выравнивании такой мотив есть у 48 последовательностей и,
кроме того, самые консервативные колонки являются общими для нашей группы и всех остальных.
Из этого можно сделать вывод, что это не новый мотив появившийся на пустом месте, а поменялись некоторые не настолько
консервативные аминокислоты и стали характерны для кроновой группы.
Рис.5 Выравнивание кроновой группы seed. Выделен найденный мотив.
PSI-BLAST
Мной был выбран P17265 (HPF_RHIME) - фактор инициации гибернации хромосом. Он распространён у прокариот,
необходим для димеризации 70s рибосом в 100s, деактивируя их, что в последствии ингибирует трансляцию.
Было запущено пять итераций PSI-BLAST(см. Таб. 1).
Начиная со второй итерации находок ниже порога вообще не было, а из описаний находок понятно, что все они
принадлежат семейству HPF.
Номер итерации
Число находок выше порога (0,005)
Идентификатор худшей находки выше порога
E-value этой находки
Идентификатор лучшей находки ниже порога
E-value этой находки
1
17
POA147.1
7.00E-04
P26983.1
0.028
2
27
P33987.1
1.00E-08
NaN
NaN
3
28
P9WMA8.1
0.002
NaN
NaN
4
28
P24694.1
3.00E-18
NaN
NaN
5
28
P24694.1
2.00E-18
NaN
NaN
Поиск мотивов де-ново для белков с доменом PF13655(RVT_N).
Из за того что для конкретной доменной структуры белки получились слишком консервативными, я воспользовался
компромиссным вариантом - взять белки архей у которых есть ещё домен PF00078(RUT_1). Затем убрал практические
идентичные и получил 99 последовательностей, на которых запускал MEME.
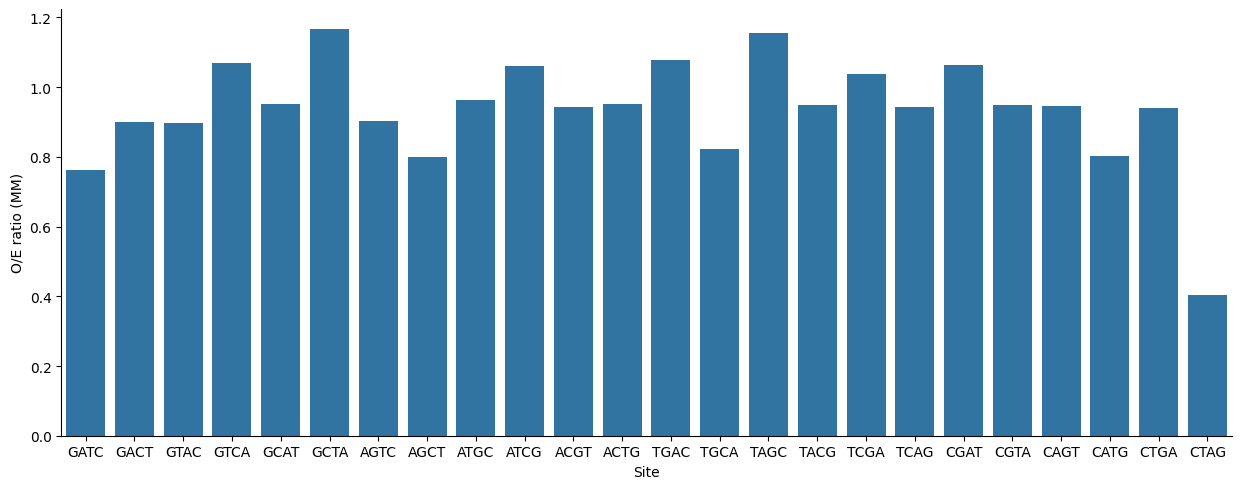
Предварительно сгенерировав 24 варианта перестановок GATC, методом Карлина подсчитаем отношение фактической
представленности к ожидаемой для каждого сайта в геноме бактерии Shigella flexneri 2a str.301.
Далее при помощи языка программирования python была построена столбчатая диаграмма:
Рис.7 Гистограмма CB для разных предполагаемых сайтов метилирования.
Наблюдается очевидная недопредставленность GATC в геноме, что не удивительно учитывая, что GATC вносит нестабильность в
цепь ДНК ввиду своей комплиментарности самому себе. Как на представленность влияют его свойства как сайта метилирования не
совсем очевидно. Но ели обратить внимание на представленность CTAG, который тоже себе комплиментарен, можно предположить, что
роль GATC как сайта метилирования положительно влияет на его поеазатель CB.