|
Главная
I Семестр
II Семестр
III Семестр
IV Семестр
V Семестр
VI Семестр
Проекты
Обратная Связь
|
Молекулярная динамика биологических молекул в GROMACS
В качестве варианта работы было выбрано:
моделирование поведения короткого пептида в формальдегиде.
Проведение моделирования
-
Были даны файлы:
- Координаты пептида: 2xl1.pdb
- Файл с ячейкой уравновешеных молекул формамида: fam_em.gro
- Файл дополнительной топологии для формамида: fam.itp
- файл праметров для минимизации энергии: em.mdp
- файл праметров для "утряски" воды: pr.mdp
- файл праметров для молекулярной динамики: md.mdp
Скачиваем их в рабочую директорию.
-
Построим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs.
pdb2gmx -f 2xl1.pdb -o pep -p pep -ff amber99sb -water tip3p -ignh
-
Сделаем небольшой отступ в ячейке от пептида.
editconf -f pep.gro -o pep_ec -d 1.5
-
Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле.
grompp -f em -c pep_ec -p pep -o pep_em -maxwarn 1
mdrun -deffnm pep_em -v 2> max_force.txt
В ходе оптимизации геометрии максимальная сила изменялась в пределах порядков 2 и 3.
Начальное значение 4.37039e+03, конечное 3.210e+02.
-
Добавим в ячейку молекулы формамида.
genbox -cp pep_em -p pep -cs fam_em.gro -o pep_s
В выводе программы указано, что добавленно 902 молекулы формамида.
-
Теперь надо изменить в текстовом редакторе файл тополгии pep.top. После строчки:
; Include forcefield parameters
добавим #include "fam.itp"
; Include forcefield parameters
#include "fam.itp"
Добавим количество молекул формамида в запись [ molecules ]
[ molecules ]
; Compound #mols
Protein_chain_A 1
FAM 902
-
Нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion.
grompp -f em -p pep -c pep_s -o pep_si -maxwarn 1
genion -s pep_s -o pep_si -p pep -np 1
где 1 - это количество положительных ионов необходимых для нейтрализации заряда системы.
Обратный заряд (-9.9999e-01) был указан в выводе grompp
-
Проведём "утряску" формамида:
grompp -f pr -c pep_si -p pep -o pep_pr -maxwarn 1
mdrun -deffnm pep_pr -v
-
Переформатируем pep_pr.gro и pep_si.gro в pdb формат. И сравним визуально в PyMol изменеия в системах.
Молекулы на картинке слева система до утряски - строго упорядочены. Справа - после утряски, система выглядит довольно хаотичной.
-
Теперь надо скопировать файл на суперкомпьютер. Сначала зайдем на суперкомпьютер и создадим папку с фамилией и вернёмся на kodomo.
ssh skif
mkdir Kurochkin
exit
-
Копируем файлы:
scp -r * skif:Kurochkin/
-
Запускаем тестовое моделирование на суперкомпьютере.
ssh skif
cd Kurochkin
grompp -f md -c pep_pr -p pep -o pep_md -maxwarn 1
mpirun -np 16 -maxtime 5 -q test /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Ошибок нет, переходим к основному моделированию.
-
Запускаем основное моделирование на суперкомпьютере.
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Номер задачи: 241017. Просмотреть ход счёта можно в файле mdrun_mpi.out-241017.
less mdrun_mpi.out-241017
Shift+. для перехода в конец файла.
Параметры моделирования
- Силовое поле используемое при построении топологии: amber99sb.
- Заряд системы: -1. Возникновения заряда в системе связано с наличием заряженных боковых групп.
- Размер и форма ячейки: параллелепипед со сторонами (нм) 5.06500, 4.67000, 4.22100.
- Минимизация энергии (файл em.mdp):
- Модель, которой описывался растворитель
Растворителя не было.
; IMPLICIT SOLVENT ALGORITHM
implicit_solvent = No
- Утряска растворителя (файл pr.mdp):
- Для биополимеров, укажите параметр который обуславливает неподвижность биополимера:
Включает файл posre.itp с номерами неподвижных атомов в топологию.
define = -DPOSRES
- Число шагов: 10000.
- Длина шага: 0.001 пс.
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
- Быстрое вычисление электростатики по методу Particle Mesh Ewald.
coulombtype = pme
-
vdw-type = Cut-off
- Алгоритмы термостата и баростата.
- Контроль температуры термостатом Берендсена.
Tcoupl = Berendsen
- Без контроля давления. Это означает фиксированный размер ячейки.
Pcoupl = no
- Основной расчёт МД (файл md.mdp):
- Время моделирования: 7 часов 22 минут 41 секунды
- количество процессоров: 16
- эффективность масштабирования: 100%
- Длина траектории: 20 нс.
- Число шагов: 10000000.
- Длина шага: 0.002 пс
- Алгоритм интегратора: md
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий.
-
coulombtype = pme
-
vdw-type = Cut-off
- Алгоритмы термостата и баростата.
- Контроль температуры по методу "Velocity rescale".
Tcoupl = v-rescale
- Контроль давления с помощью экспоненциальной релаксации. Ячейка пересчитывается каждый шаг.
Pcoupl = Berendsen
Анализ результатов
- Начнём с визуального анализа движений молекул. Переводим траекторию в формат PDB и смотрим в PyMOL:
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_pbc_1.pdb -skip 20 -pbc mol
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_fit_1.pdb -skip 20 -fit rot+trans
Выбираем файл pep_fit_1.pdb, для визуализации так как в нем молекула пептида все время находится в центре.
Уже на втором кадре происходит небольшое изменение структуры, это модель 1 - 200 фемтосекунд от начала. Видно, как в основном C-конец альфа-спирали то расплетается, то принимает прежнее положение. Но начиная, где то с 26 кадра (25 модель - 5 наносекунд) виток α-спирале на С-конце претерпевает сильное изменение, и уже не возвращается прежнее состояние. За время моделирования пептид не расплавился на N-конце, это вероятно связано с тем, он стабилизировался за счет взаимодействий боковых груп.
- Определим средне-квадратичное отколнение в ходе моделирования. Так как у нас происходит конформационный переход сначала расчитаем отклонение в ходе все симуляции относительно стартовой структуры.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_1

Наблюдается сильный рост RMSD вначале, то есть молекула сразу уходит из своего начального состояния. А в ходе моделирования отклонение то растёт, то убывает, не опускаясь до нуля. Также видно, что RMSD не выходит за пределы 0.5, что довольно мало. Это говорит о том, что пептид несильно меняет свою конформацию, сохраняя в целом альфа-спиральную структуру.
- И относительно каждой предыдущей структуры на растоянии 400 кадров. Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_2 -prev 400
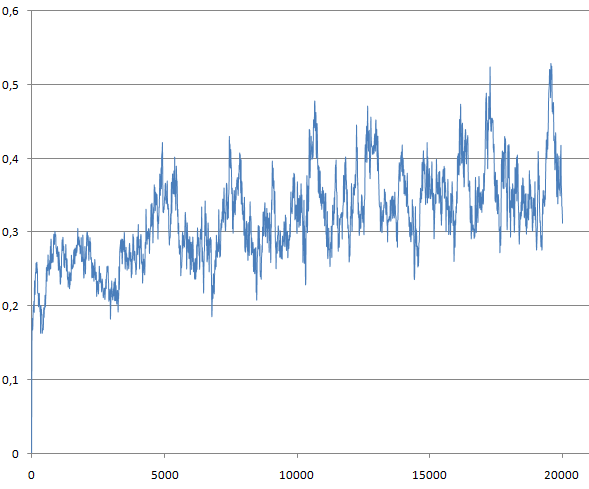
К концу моделирования данное отклонение не уменьшается, то есть молекула не принимает какую-либо устойчивую конформацию.
- Определим изменение гидрофобной и гидрофильной поверхности в ходе конформационного перехода.
g_sas -f pep_md.xtc -s pep_md.tpr -o sas_pep.xvg
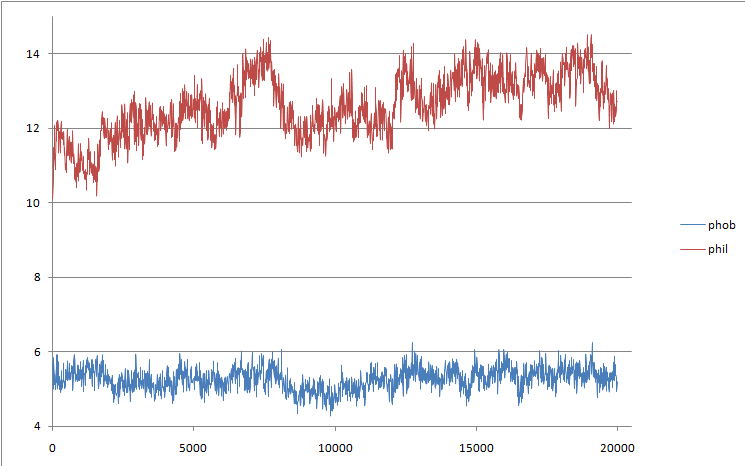
Гидрофобная поверхность практически не изменяется. Она включает в себя только гидрофобные остатки. Как они были направлены от спирали в раствор вначале, так и сохранили своё положение до конца. А гидрофильная поверхность немного увеличилась, это вероятно связано с тем, что количество связей между растворителем и пептидом увеличилось.
- Традиционным анализом является расчёт колчества образуемых водородных связей. Если мы будем исследовать связи между пептидом и пептидом, то это будут водородные связи в пептиде.
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep
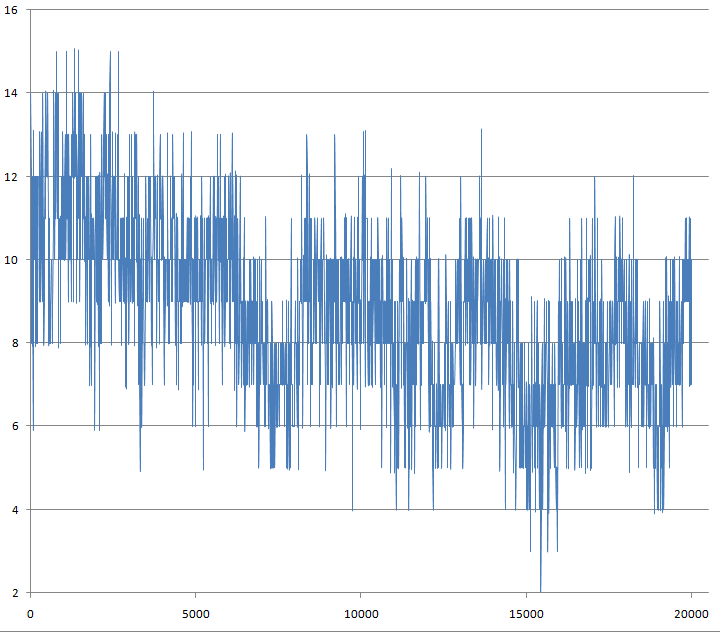
Видно, что количество водородных связей внутри пептида постоянно меняется, опускаясь даже до 2, и возрастая до 15.
- Не менее интересно будет изучить количество вдородных связей пептид-формамид.
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep_sl
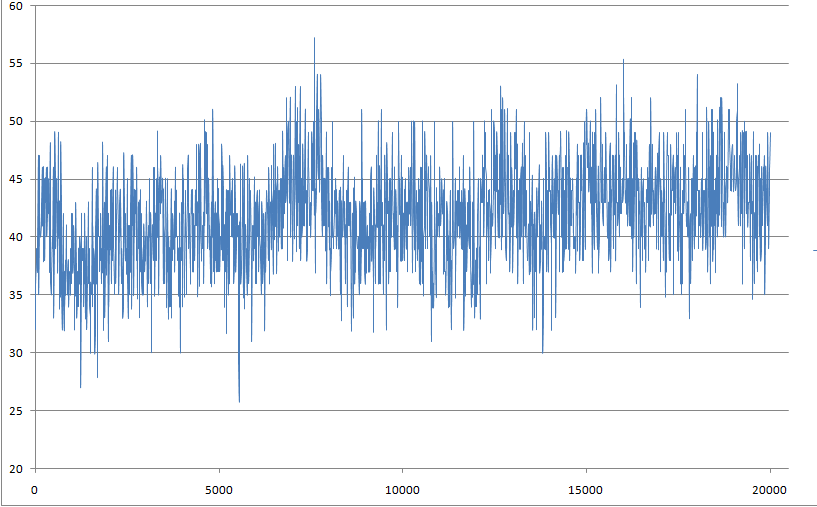
Количество водордных связей с растворителем то растёт, то убывает и не приходит к какому-то определённому значению. И в итоге получили, что количество водородных связей в конечном состояние больше, чем в начальном.
- Если у нас происходит разрушение вторичной структуры, то надо построить зависимость вторичной структуры от времени:
do_dssp -f pep_md.xtc -s pep_md.tpr -o ss
# Для просмотра переведём xpm в eps
xpm2ps -f ss.xpm -o ss.eps -by 10
# Переведём в картинку
convert ss.eps ss.png
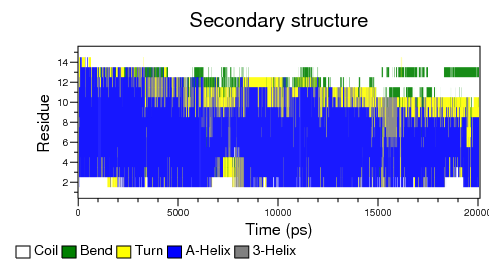
Расчёт вторичной структуры совпадает со всеми наблюдениями. Видно, что α-спираль расплелась на C-конце, а структура на N-конце практически не изменялась в процессе молекулярной динамики.
|