|
Главная
I Семестр
II Семестр
III Семестр
IV Семестр
V Семестр
VI Семестр
Проекты
Обратная Связь
|
Cеми-эмпирические вычисления: Mopac
Установка переменных:
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Оптимизация структуры порфирина
Создадим файл с аннотацией порфирина в виде SMILES. Подадим его на вход программе obgen для построения 3D структуры порфирина: obgen 1.smi > 1.mol. Просмотрим полученную структуру в PyMol и удалив ненужные водороды, получаем 3D-структуру порфирина.
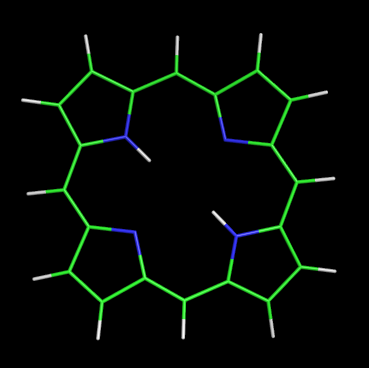
Сбоку молекула порфирина выглядит так:
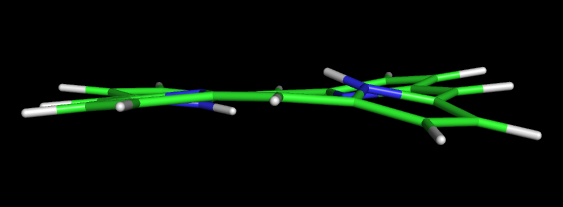
Как видно, построенная молекула не является плоской, хотя должна быть такой.
Программой babel получаем входные файлы для программы Mopac с параметризацией PM6 и AM1 и запускаем Mopac:
MOPAC2009.exe file_name. И программой babel переводим выходной файл с расширением .out в PDB. Сравним полученные оптимизации.
Обе параметризации дали неплохой результат, структуры выглядят плоскими. Но с AM1 Mopac справился немного получше.
Возбужденные состояния порфирина
Для указания Mopac о необходимости расчёта возбуждённого состояния добавляем в конец файла:
пустая строка
cis c.i.=4 meci oldgeo
some description
Рассмотрим выходной файл porphyrin_hesit.out и найдем в нем значения энергий для электронных переходов:
STATE ENERGY (EV) Q.N. SPIN SYMMETRY POLARIZATION
ABSOLUTE RELATIVE X Y Z
1+ 0.000000 0.000000 1+ SINGLET ????
2 1.913312 1.913312 1 TRIPLET ????
3 2.266014 2.266014 2 SINGLET ????
4 2.463186 2.463186 2 TRIPLET ????
5 2.823915 2.823915 3 TRIPLET ????
6 3.362161 3.362161 4 TRIPLET ????
7 3.389757 3.389757 3 SINGLET ???? 0.2031 0.2347 0.0010
8 3.669242 3.669242 4 SINGLET ???? 2.3899 2.0438 0.0085
9 3.871323 3.871323 5 SINGLET ???? 1.5461 1.7992 0.0084
The "+" symbol indicates the root used.
На основании этих значений энергии рассчитаем длины волн, при которых происходят эти переходы:
| Энергия (eV) |
Длина волны (nm) |
| 1.913312 |
649 |
| 2.266014 |
548 |
| 2.463186 |
504 |
| 2.823915 |
440 |
| 3.362161 |
369 |
| 3.389757 |
366 |
| 3.669242 |
338 |
| 3.871323 |
321 |
Оптимизация парабензохинона
Для молекулы O=C1C=CC(=O)C=C1 (парабензохинон) была определена геометрия с помощью obgen (quinine_obgen.pdb) и Mopac, использовалась параметризацию pm6 (quinine_neutral.pdb).
obgen (голубые углероды) и нейтральная молекула в Mopac (желтые углероды):
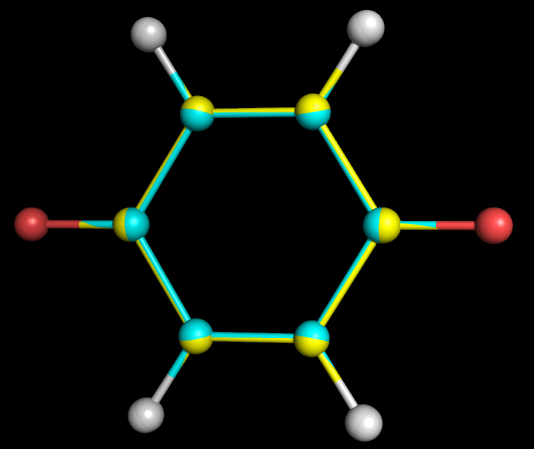
Видно, что структуры различаются незначительно.
Затем была проведена оптимизация для дианиона этой молекулы. В первую строчку mop файла добавляем слово CHARGE=-2 и явным способом указываем (последовательность (-) справа от имени атома), что отрицательный заряд должен находиться на кислородах. В результате получили: quinine_dianion.pdb
obgen (голубые углероды) и дианион в Mopac (коричневые углероды):
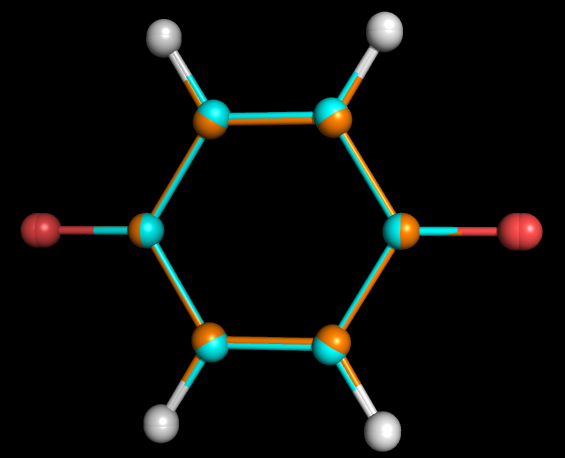
Из наложения видно, что в дианионе структура претерпевает некоторые изменения. Связи С-O удлиняются, так как из двойных они становятся одинарными, к тому же цикл становится ароматическим.
|

