|
Главная
I Семестр
II Семестр
III Семестр
IV Семестр
V Семестр
VI Семестр
Проекты
Обратная Связь
|
Изучение работы методов контроля температуры в GROMACS
-
Начнем с того, что подготовим файл координат и файл топологии. В прошлом занятии был предоставлен gro файл с 38 молекулами этана. Создадим индекс файл в котором будет группа из одной молекулы этана.
make_ndx -f box_38.gro -o 1.ndx
Выбераем остаток номер 1. Появляется новая группа.
Теперь создадим gro файл с одной молекулой и зададим ячейку. При запуске editconf выбераем номер, соответствующей группе из одной молекулы.
editconf -f box_38.gro -o et1.gro -n 1.ndx
#зададим ячейку и расположим молекулу по центру ячейку
editconf -f et1.gro -o et.gro -d 2 -c
Получили файл et1.gro. Исправим файл топологии et.top из прошлого задания. В разделе [ molecules ] изменим количество молекул этана с 38 на 1.
-
Даны 5 файлов с разными параметрами контроля температуры:
- an.mdp - метод Андерсена для контроля температуры.
- be.mdp - метод Берендсена для контроля температуры.
- nh.mdp - метод Нуза-Хувера для контроля температуры.
- sd.mdp - метод стохастической молекулярной динамики.
- vr.mdp - метод "Velocity rescale" для контроля температуры.
Напишем скрипт (make.bash) для работы с 5ю системами.
-
Создаём отдельную папку для каждого метода.
mkdir ${i}
# где i: an,be,nh,sd,vr
Сначала надо построить входные файлы для молекулярно-динамического движка mdrun с помощью grompp:
grompp -f ${i}.mdp -c et.gro -p et.top -o et_${i}.tpr
-
У нас получилось 5 tpr файлов. Теперь для каждого из них запустим mdrun.
mdrun -deffnm et_${i} -v -nt 1
-
Теперь переходим к анализу результатов. Начнем с визуального анализа. Для каждой из 5 систем проведем конвертацию в pdb и просмотрим в PyMol.
trjconv -f ${i}/et_${i}.trr -s ${i}/et_${i}.tpr -o ${i}/et_${i}.pdb
Полученные ролики для PyMOL:
Некоторые наблюдения:
- В методе Андерсена наблюдаются небольшие колебания по длинам связей и валентным углам. Вращение в данном случае отсутствует. Такое состояние, вероятно, характерно для кристалла.
- В методе Берендсена сперва мы наблюдать различные колебания и вращения, но затем молекула постепенно начинает всё больше вращаться, а амплитуда колебаний уменьшается.
- В методе Нуза-Хувера мы наблюдаем небольшие колебания и вращение по связи С-С. Молекула в основном находится в заторможенной конформации, но также видна и заслонённая.
- В методе стохастической молекулярной динамике молекула очень быстро вращается и перемещается. Затруднительно проследить за направлением движения молекулы, как будто она случайным образом меняет свою ориентацию или сталкивается с другими молекулами.
- Метод "Velocity rescale" напоминает метод Нуза-Хувера, но в данном случае мы наблюдаем более амплитудные колебания и уменьшенное вращение по связи С-С. Молекула в основном находится в заторможенной конформации, крайне редко прослеживаются конформации близкие к заслонённой.
-
Сравним потенциальную энергию связи и кинетическую энергию для каждой из 5 систем.
echo -e "Bond\nKinetic-En.\n0" | \
g_energy -f ${i}/et_${i}.edr -o ${i}/et_${i}_en.xvg > ${i}/et_${i}_en.txt
Построим графики изменения энергий. Для этого создадим файлы скриптов для Gnuplot и выполним их (получившиеся графики сохраним в формате png):
echo -e "set datafile commentschars '#@&'
set term 'png'
set output '${i}/en_${i}.png'
plot '${i}/et_${i}_en.xvg' using 1:2, '${i}/et_${i}_en.xvg' using 1:3" > ${i}/en_${i}.gnu
gnuplot < ${i}/en_${i}.gnu
-
Рассмотрим распределение длинны связи С-С за время моделирования. Сначала создадим индекс файл с одной связью. В текстовом редакторе создим файл b.ndx со следующим содержимым:
[ b ]
1 2
И запустим утилиту по анализу связей g_bond:
g_bond -f et_${i}.trr -s et_${i}.tpr -o bond_${i}.xvg -n b.ndx
Построим графики распределения длинн связей. Для этого создадим файлы скриптов для Gnuplot и выполним их (получившиеся графики в виде boxes сохраним в формате png):
echo "set datafile commentschars '#@&'
set term 'png'
set output '${i}/bond_${i}.png'
plot '${i}/bond_${i}.xvg' with boxes" > ${i}/bond_${i}.gnu
gnuplot < ${i}/bond_${i}.gnu
Мы хотим получить одно изображения.
Для этого сперва получим список изображений:
ls -r ${i}/*.png >> image_list
А затем вопользовавшись программой montage cольём все изображения в одно.
montage +frame +shadow +label -tile 2x5 -geometry 700x500 `cat image_list` temp_images.png
В итоге получаем следующее изображение. На изображениях слева энергия для каждого состояния: красным цветом потенциальная энергия связей, зелёным - кинетическая энергия; справа распределения длин связей:
| AN |
 |
| BE |
| NH |
| SD |
| VR |
-
Распределение Максвелла-Больцмана имеет следующую форму:
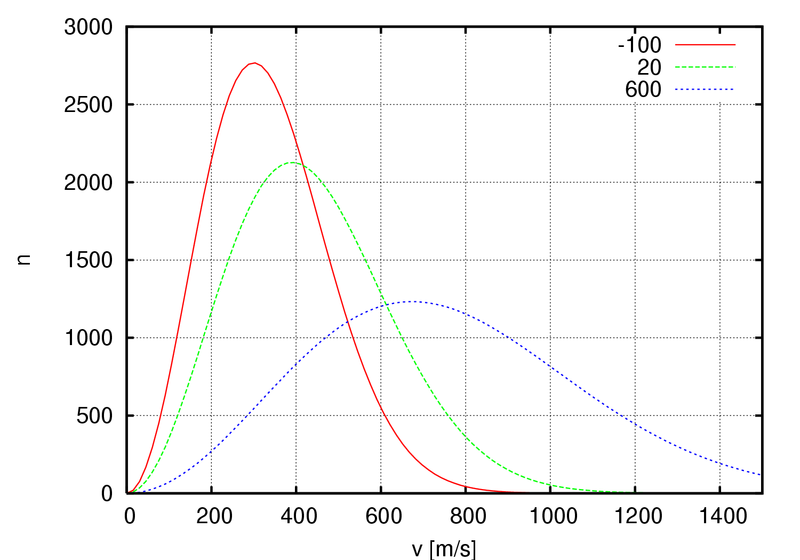
Ему совершенно не соответствуют графики методов Андерсена и Берендсена, неплохо соответствуют графики методов стохастической молекулярной динамики и "Velocity rescale".
Исходя из всех наблюдений, можно сказать, что реалистичны 2 метода: стохастической молекулярной динамики и "Velocity rescale". Но на роликах в PyMOL метод стохастической молекулярной динамики кажется немного странным. Поэтому, на мой взгляд, метод "Velocity rescale" является наиболее реалистичным для поддержания температуру в системе.
|