| FastQC | Анализ качества чтений. | fastqc chr15.2.fastq |
| Trimmomatic | Очистка чтений | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr15.2.fastq chr15_new.fastq TRAILING:20 MINLEN:50 |
| hisat2-build | Индексирование референсной последовательности | hisat2-build chr15.fasta chr15 |
| hisat2 с параметром --no-softclip | Выравнивание прочтений и референса в формате .sam. | hisat2 -x chr15 chr15_new.fastq --no-softclip >> alignment.sam |
| samtools view | Перевод выравнивания чтений с референсом в бинарный формат .bam | samtools view alignment.sam -b >> alignment.bam |
| samtools sort | Сортировка выравнивания чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения | samtools sort alignment.bam -T alignment.txt -o sort.bam |
| samtools index | Индексирование отсортированный .bam файл | samtools index sort.bam |
| samtools idxstats | Выдача статистики в индексном файле, соответствующем входному файлу. | samtools idxstats sort.bam >> idxstats.txt |
| htseq-count | Подсчет числа чтений по определённому критерию | htseq-count -f bam -s no sort.bam /P/y14/term3/block4/SNP/rnaseq_reads/ gencode.v19.chr_patch_hapl_scaff.annotation.gtf >> htseq-count.txt |
1. Анализ качества чтений.
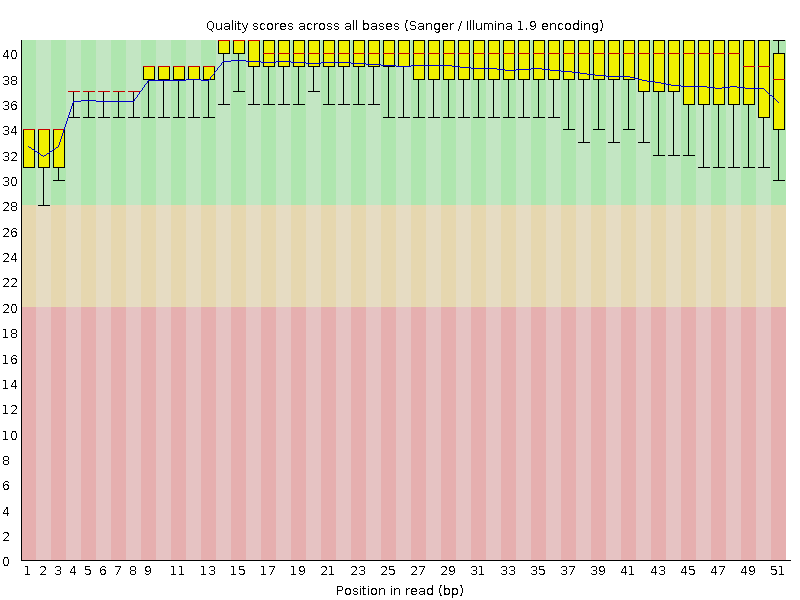
Для выполнения данного задания взят файл chr20.1.fastq. С помощью программы FastQC проанализируем качество чтений.
Общее число последовательностей составляет 3565, длина составляет 41-51.
Чтения хорошего качества, поэтому применение программы trimmomatic не требуется.
| gene_id | Количество ридов | Тип гена | Имя гена | Белок (UniProt) |
| ENSG00000125835.13 | 2110 | Protein coding | SNRPB | small nuclear ribonucleoprotein polypeptides B and B1 |
| ENSG00000251806.1 | 4 | SnoRNA | SNORD119 | - |
© Наумова Юлия, 2018