|
|
|
|
|
|
|
|
|
|
|
 |
|
||||||||
|
|
|
|
|
|
|
|
|
||
|
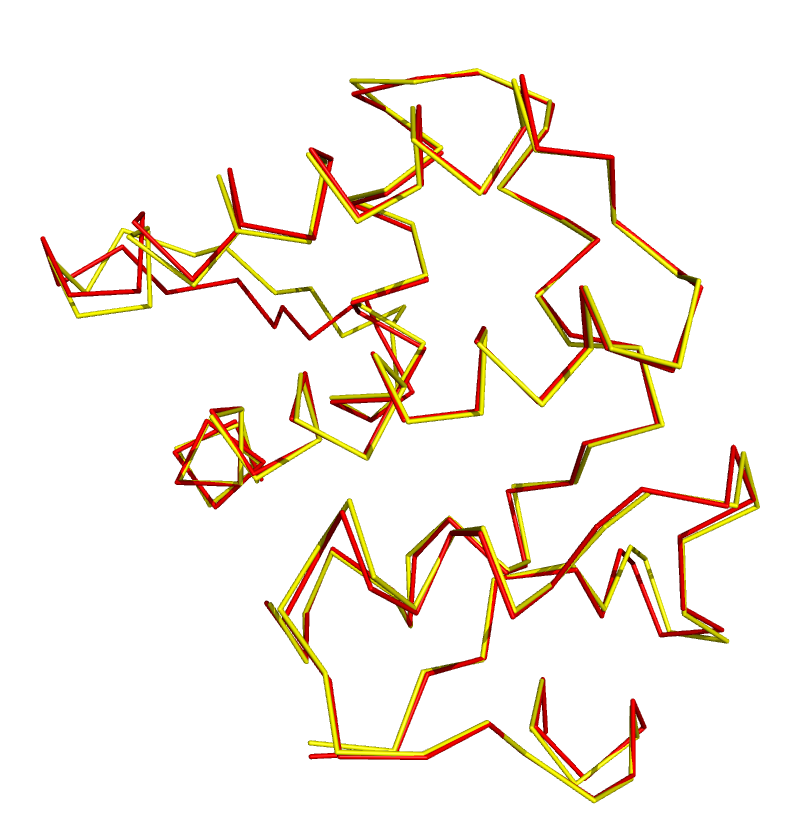
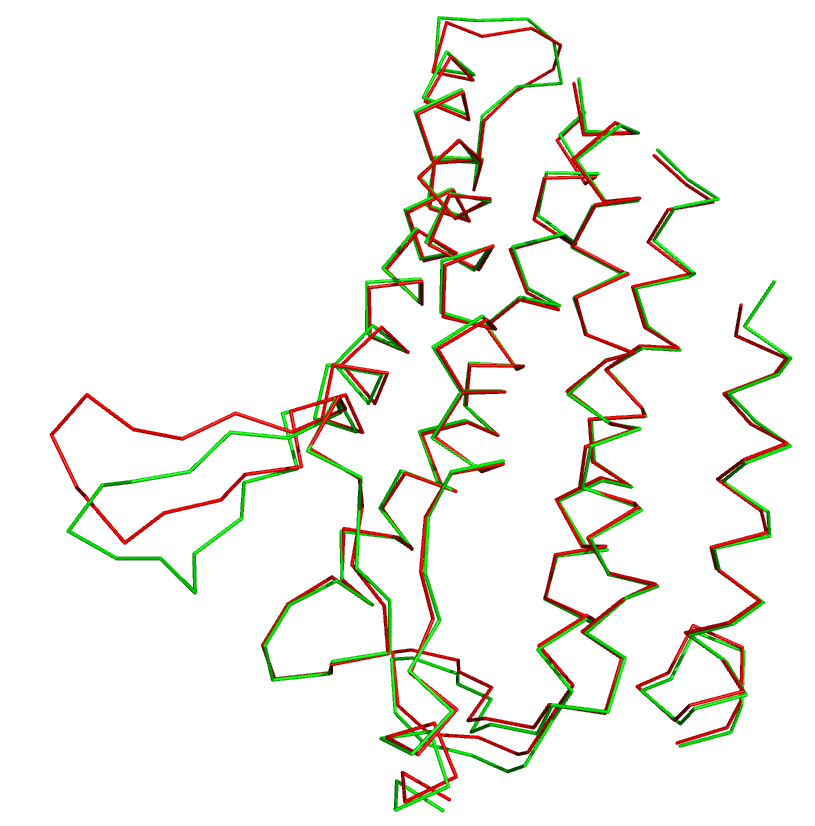
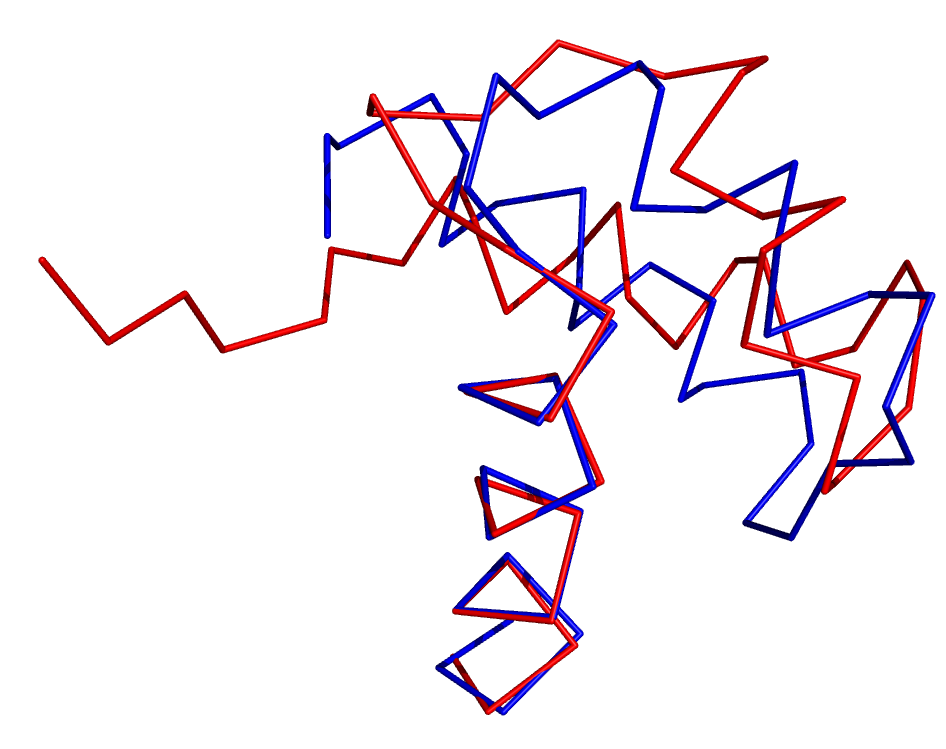
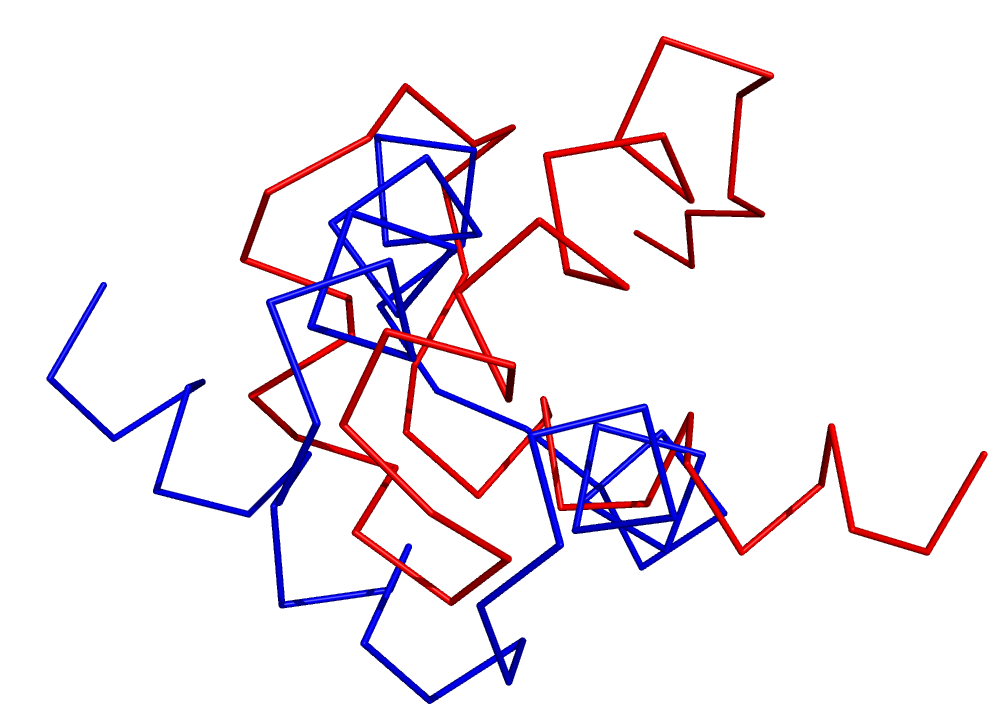
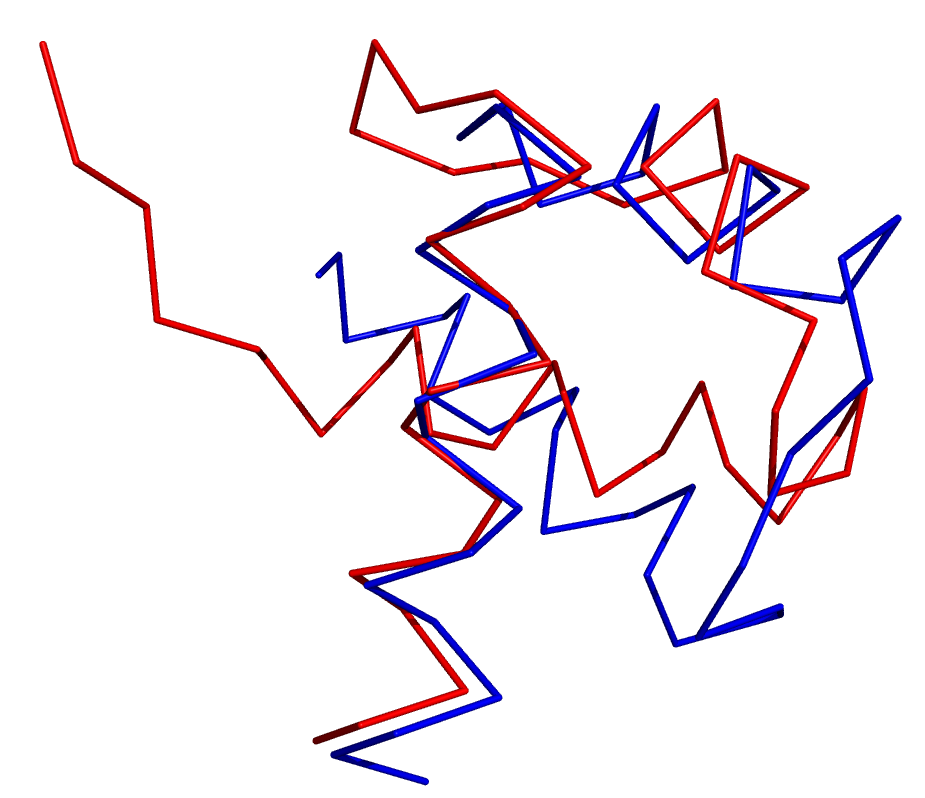
Пространтсвенное выравнивание и совмещение Построил в PyMOL командой align три совмещения следующих структур: цепи A из записи 1HY0 и цепи A из записи 1I0A. Алгоритм CATH выделяет три домена в данной цепи, которые были совмещены: 1) 27-123 , 195-234 RMS = 0.371 (773 to 773 atoms)
RMS = 0.409 (1371 to 1371 atoms)
RMS = 0.527 (449 to 449 atoms)
На данных картинках видно консервативность доменов пространственной структуры цепи А двух белков, это видно и из RMS, которое в 3 случаях мало, что говорит о большой схожести пространственных структур.
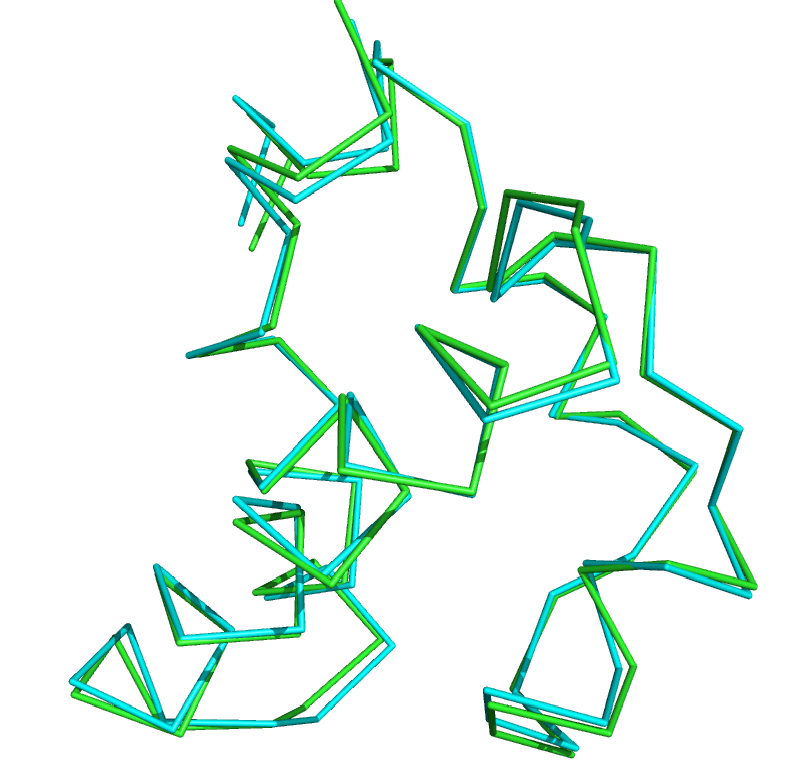
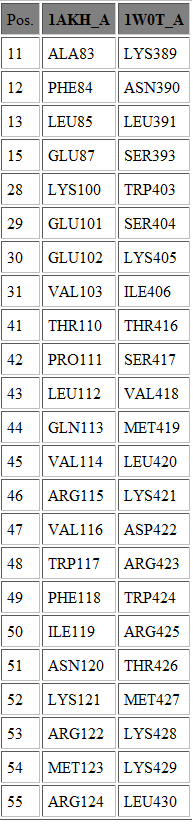
Построил программой PDBeFOLD (она же SSM) структурное выравнивание цепи A из 1AKH и цепи A из 1W0T: >akh:A >w0t:A
По данному выравниванию с помощью сервиса Geometrical core найшел геометрические ядра с порогом 2 Å. Совместил в PyMOL командой pair_fit структуры по CA-атомам, входящим в геометрическое ядро.
RMS = 0.406 (15 to 15 atoms) данного совмещения мал, что говорит о высокой схожести цепи данных двух белков Сравнил с результатом команды align.
RMS = 6.198 (161 to 161 atoms) Высокий RMS говорит о плохом совмещении цепей. Сравнил с результатом экспериментальной команды super.
RMS = 2.186 (192 to 192 atoms) RMS данного совмещения меньше, чем при команде align, но выше чем через построение через геометрические ядра.
|
| ||||||||
|
|
|||||||||
|
© Замараев Алексей |
|
||||||||