Вычисление параметров для молекулярной механики
Для вычисления параметров для молекулярной механики была предоставлена оптимизированная структура этана в виде z-matrix:
$DATA
eth
C1
C
C 1 cc
H 2 ch 1 cchv
H 2 ch 1 cch 3 d1 0
H 2 ch 1 cch 3 d2 0
H 1 ch 2 cch 3 d3 0
H 1 ch 2 cch 5 d3 0
H 1 chv 2 cch 4 d3 0
cc=1.52986
ch=1.08439
chv=1.08439
cch=111.200
cchv=111.200
d1=120
d2=-120
d3=180
$END
Наша цель состоит в том, что бы создать порядка 20 разных файлов для расчёта энергии в Gamess с разными значениями по длине одной из связей.
Файл-заготовка для размножения - et.inp. Для этого к координатам добавьте шапку для dft из предыдущего практикума. Только надо изменить информацию о типе входных координат: замените COORD=CART на COORD=ZMT.
Создадим скрипт, который будет создавать необходимые файлы и выполнять расчёт с помощью GAMESS со значениями энергии для каждого файла.
make_b.bash
Запускаем скрипт командой и сразу же перенаправляем выход в файл bond.txt:
bash ./make_b.bash > bond.txt
У нас есть зависимость энергии молекулы от длины одной связи. Эту зависимость можно построить в gnuplot.
Запустите Xming->XLaunch.
Перейдите в рабочую директорию. Запустите Gnuplot:
gnuplot
Построим зависимость энергии от длины связи, просто введем : plot "bond"
У нас должен появился график с точками похожими на параболу. Теперь нам надо найти коэффициенты в функции f(x)=a+k(x-b)^2 которые бы позволили наиболее близко описать наблюдаемую зависимость. Для этого воспользуемся возможностями Gnuplot. Сначала зададим функцию в развернутом виде, в строке gnuplot введём:
f(x)=a + k*x*x - 2*k*x*b + k*b*b
И зададим стартовые значения коэффициентов:
a=-80
k=1
b=1.5
Проведём подгонку коэффициентов под имеющиеся точки в файле bond:
fit f(x) "bond" via a,k,b
Значения коэффициентов
a = -79.7652
k = 0.563608
b = 1.55432
Построим графики функции и значений энергии из Gamess.
plot "bond", f(x)
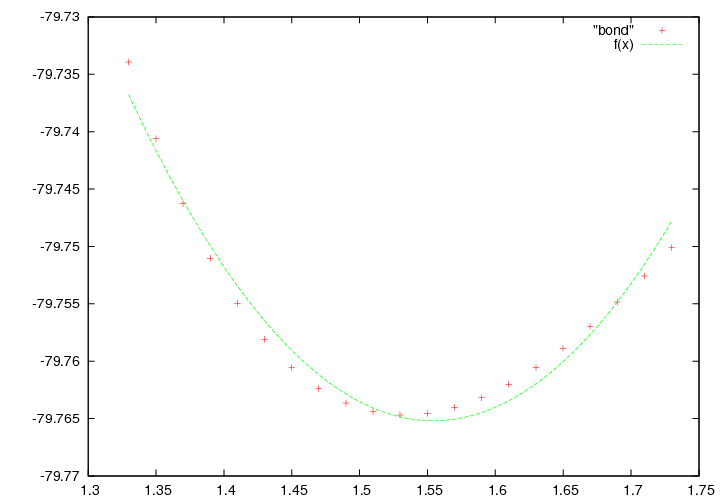
Функция неточно совпадает с точками, так как в данном случае мы наблюдаем участок потенциала Мориса, имеющий более сложную зависимость.
Сделаем аналогичную работу для валентного угла HCC, его значения должны изменяться от 109.2 до 113.2. Апроксимируем зависимость так же параболой:
f(x)=a + k*x*x - 2*k*x*b + k*b*b
Скрипт: make_v.bash
Энергии: valent_ener.txt
Значения коэффициентов:
a = -79.7647 +/- 1.207e-08 (1.513e-08%)
k = 3.56075e-05 +/- 6.214e-09 (0.01745%)
b = 111.38 +/- 9.931e-05 (8.916e-05%)
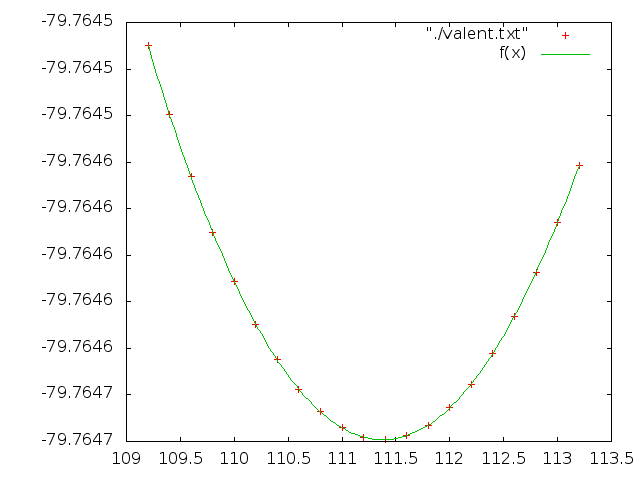
Как видно, апроксимация достаточно хорошая.
Сделаем тоже самое и для торсионного угла d3, его значения должны изменяться от -180 до 180 c шагом 12. Зависимость в данном случае апроксимируется косинусом:
f(x)=a*cos(k*x/180*pi) + b
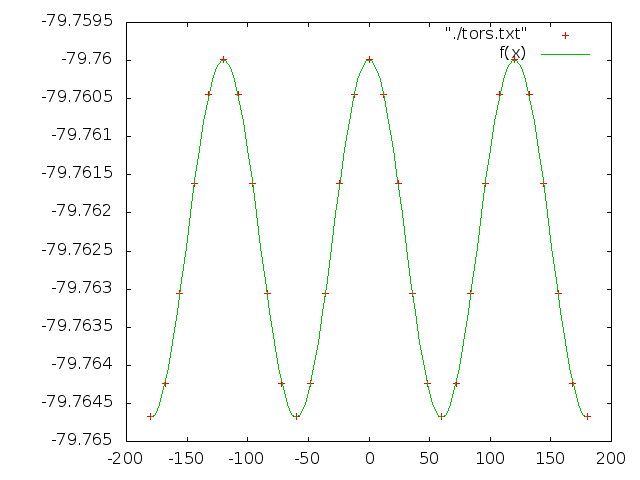
Энергии: tors.txt
Значения коэффициентов:
a = 0.00234519 +/- 8.891e-07 (0.03791%)
k = 3.00014 +/- 0.0002247 (0.007491%)
b = -79.7623 +/- 6.577e-07 (8.245e-07%)
|
