Часть I: подготовка чтений
Команды:
| fastqc chr6.fastq | Анализ качества чтений, получаем chr6_fastqc.html файл с отчетом |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6.fastq chr6_2.fastq SLIDINGWINDOW:10:20 | Проходит по каждому чтению окном длиной в 10 нуклеотидов, как только среднее качество в окне становится ниже, чем 20, удаляет все, что правее окна |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6_2.fastq chr6_2_cleaned.fastq MINLEN:50 | Удаляет все чтения длиной меньше 50 оснований |
| fastqc chr6_2_cleaned.fastq | Анализ качества чтений, получаем chr6_2_cleaned_fastqc.html файл с отчетом |
| grep -c + chr6_2_cleaned.fastq | Считает число чтений в chr6_2_cleaned.fastq |
| grep -c + chr6.fastq | Считает число чтений в chr6.fastq |
| До | После |
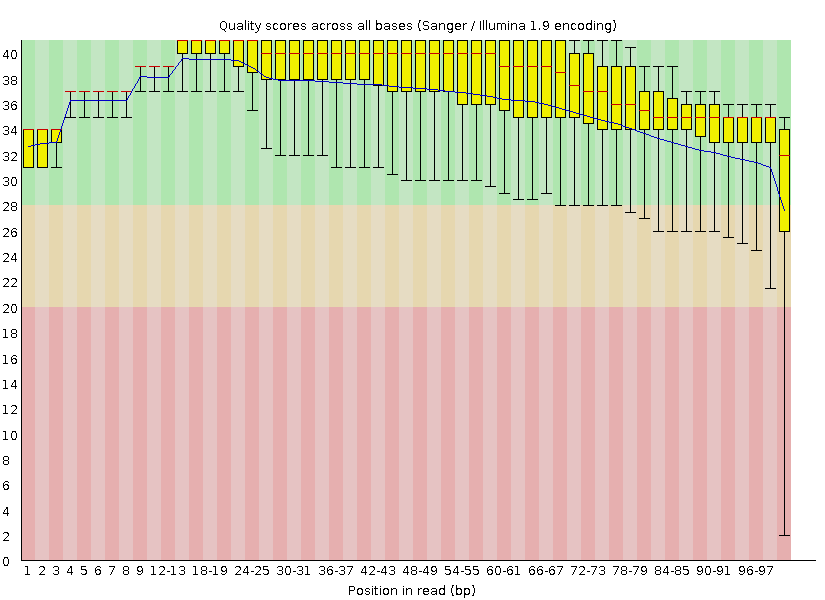 |
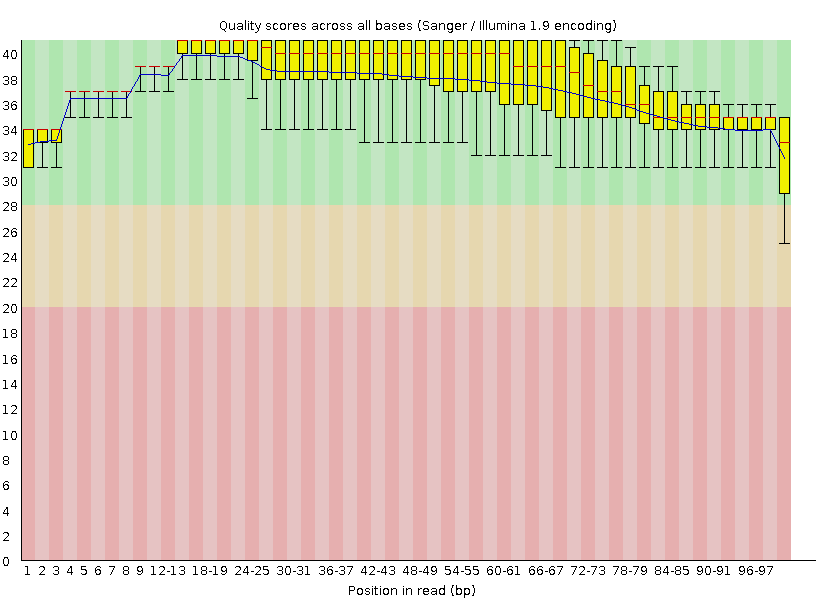 |
До чистки было 10865 чтений, а после - стало 10208. Отсеялись целиком все чтения, длина которых - меньше 50 нуклеотидов , и отрезались у некоторых чтений с правого конца фрагменты, у которых в окне длиной 10 нуклеотидов среднее качество стало ниже 20. За исключением правых концов картинки различаются мало.
| До | После |
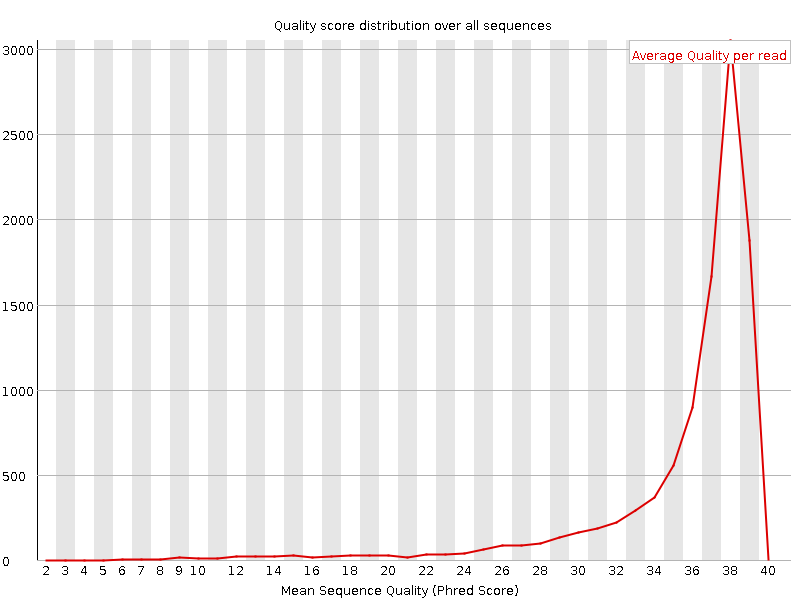 |
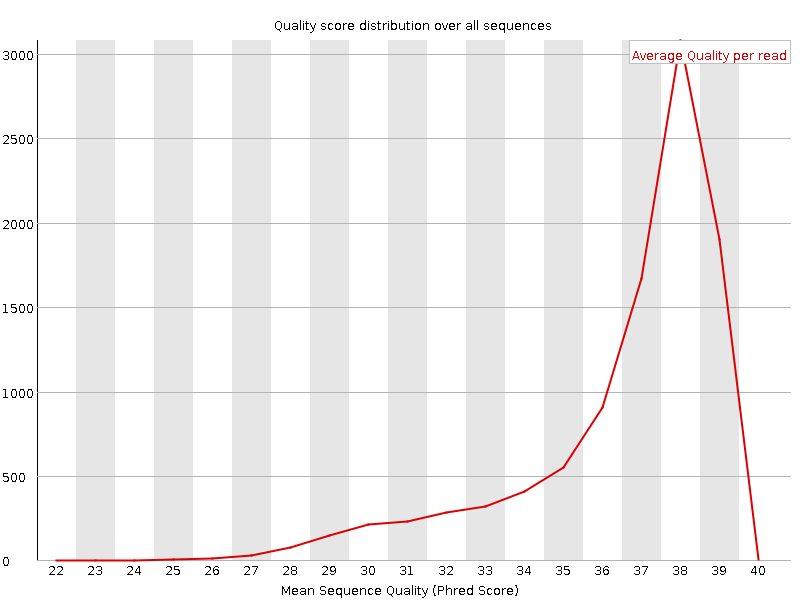 |
На этих картинках распределение ридов по качеству. Видно, что после чистки среднее не изменилось, но утяжелился хвост распределения.
Часть II: картирование чтений
Команды:
| hisat2-build chr6.fasta chr6_indexed | Индексирует референсную последовательность chr6.fasta и записывает результат в chr6_indexed. |
| hisat2 --no-spliced-alignment --no-softclip -x chr6_indexed -U chr6_2_cleaned.fastq -S chr6_aligned.map | Выравнивает риды на референс и записывает результат в chr6_aligned.map. |
| samtools view -bS chr6_aligned.map > chr6_aligned.bam | Переводит выравнивания из формата .map в формат .bam, результат - в файле chr6_aligned.bam. |
| samtools sort -T ./tmp/sorted.tmp -o chr6_aligned_sorted.bam -O bam chr6_aligned.bam | Сортирует выравнивания по координате начала чтения в референсе. Результат - в файле chr6_aligned_sorted.bam. |
| samtools index chr6_aligned_sorted.bam | Индексирует отсортированный .bam файл. |
Вывод hiset2 выглядит следующим образом:
|
9879 reads; of these: 9879 (100.00%) were unpaired; of these: 64 (0.65%) aligned 0 times 9815 (99.35%) aligned exactly 1 time 0 (0.00%) aligned >1 times 99.35% overall alignment rate |
Из 9879 ридов, содержащихся в файле chr6_2_cleaned.fastq 99.35%, то есть 9815 ридов были откартированы ровно 1 раз на референсную последовательность хромосомы; 0.65% (64 рида) не откартировались совсем, а больше одного раза ни один рид не откартировался.
Часть III: Анализ SNP
Команды:
| samtools mpileup -uf chr6.fasta chr6_aligned_sorted.bam -o chr6_snp.bfc | Создает файл с полиморфизмами в формате .bcf |
| bcftools call -cv chr6_snp.bfc -o chr6_snp.vcf | Создает файл со списком отличий между референсом и чтениями в формате .vcf |
| convert2annovar.pl chr6_snp.vcf -format vcf4 -outfile chr6_snp.annovar | Переводит .vcf файл в формат annovar. |
| cat chr6_snp.annovar | grep -v "-" > chr6_snp_only.annovar | Оставляет в файле только snp. |
| annotate_variation.pl -out ngs/zhenia/chr6_snp_only_refgene -build hg19 ngs/zhenia/chr6_snp_only.annovar annovar/humandb/ | Аннотирует полученные snp по базe данных refgene. |
| annotate_variation.pl -filter -out ngs/zhenia/chr6_snp_only_dbsnp -build hg19 -dbtype snp138 ngs/zhenia/chr6_snp_only.annovar annovar/humandb.old | Аннотирует полученные snp по базe данных dbsnp. В файл dropped попадают проаннотированные snp, а в файл filtered - непроаннотированные. |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out ngs/zhenia/chr6_snp_only_1000 ngs/zhenia/chr6_snp_only.annovar annovar/humandb.old/ | Аннотирует полученные snp по базe данных 1000 genomes. |
| annotate_variation.pl -regionanno -build hg19 -out ngs/zhenia/chr6_snp_only_gwas -dbtype gwasCatalog ngs/zhenia/chr6_snp_only.annovar annovar/humandb.old/ | Аннотирует полученные snp по базe данных GWAS. |
| annotate_variation.pl ngs/zhenia/chr6_snp_only.annovar annovar/humandb.old/ -filter -dbtype clinvar_20150629 -buildver hg19 -out ngs/zhenia/chr6_snp_only_ clinvar | Аннотирует полученные snp по базe данных Clinvar. |
Описание полиморфизмов:
| Тип | Координата | Было | Стало | Покрытие | Качество |
| INDEL | 107006115 | TCCCCC | TCCCC,TCCC | 5 | 15.1077 |
| Замена | 106961119 | T | C | 6 | 36.0297 |
| INDEL | 154344146 | tc | tCc | 1 | 3.25208 |
В .vcf файле 6 инделей и 76 snp. Средняя глубина покрытия у snp - 21, хотя варьирует от 1 до 169; среднее качество - 106, варьирует на два порядка. База данных refseq делит snp: по расположению в экзонной/интронной/нетранслируемой областях - 15/60/3 snp соответственно; по расположению в гене CRYBG1/TNFAIP3/OPRM1 - 43/8/27 snp; по нахождению в гомо/гетерозиготе - 42/36 snp. Cсылка на таблицу с нуклеотидными и аминокислотными экзонными snp. rs имеют 73 snp. Частота встречаемости в популяции найденных snp варьирует от 0.00658946 до 0.941094, среднее - 0.454245787. Клиническую аннотация имеет только 1 snp: замена T на G в экзоне гена TNFAIP3 с качеством 225.009 и глубиной прочтения 46. Идентификатор в базе ClinVar CN169374, но , похоже, экспериментально ничего интересного не получено. Сводная таблица по всем базам данных, полученная скриптом.