1. Анализ качества чтений
Команды:
| fastqc chr6.1.fastq | Анализирует качество чтений программой fastqc |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6.1.fastq chr6cleaned.1.fastq LEADING:28 | Убирает из начала чтения нуклеотиды качества ниже 28 |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr6cleaned.1.fastq chr6_cleaned.1.fastq MINLEN:50 | Удаляет риды длиной меньше 50 |
| До | После |
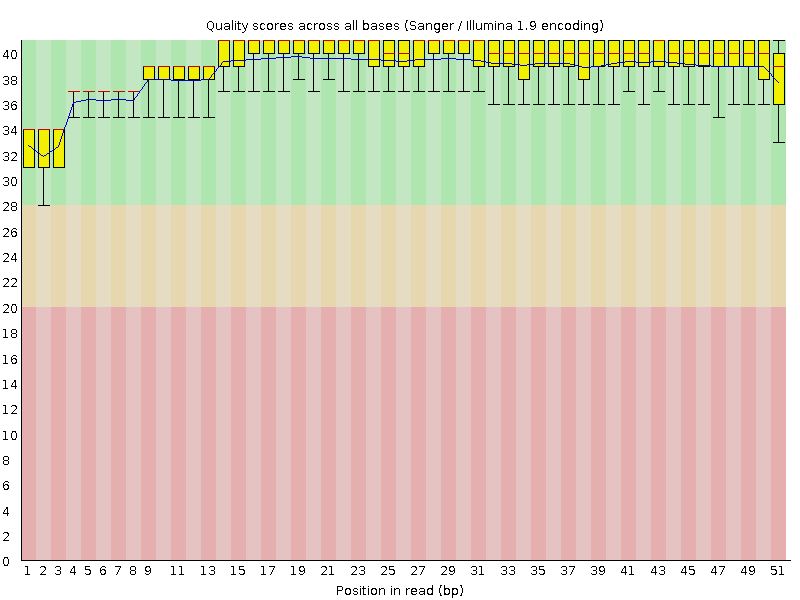 |
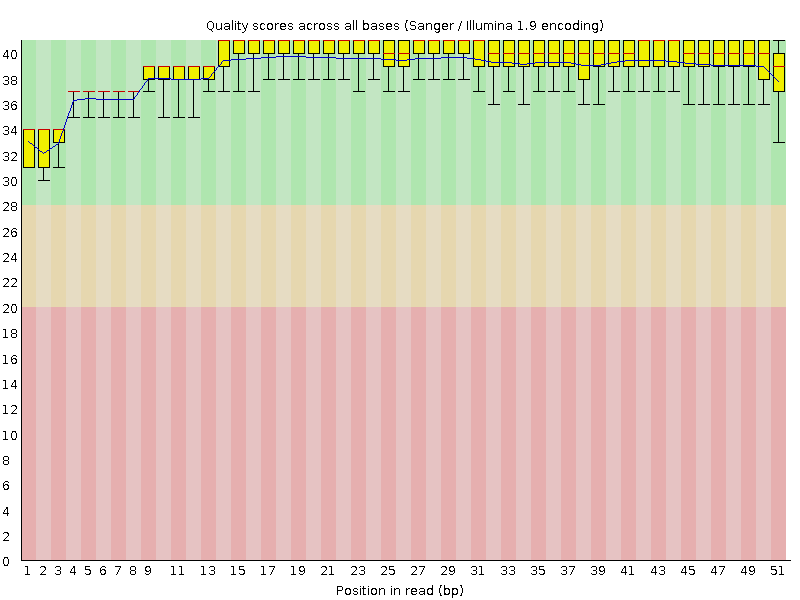 |
Количество ридов до чистки: 56575, после: 54997.
2. Картирование чтений
| hisat2-build chr6.fasta chr6_indexed | Индексирует референсную последовательность chr6.fasta и записывает результат в chr6_indexed |
| hisat2 --no-softclip -x chr6_indexed -U chr6_cleaned.1.fastq -S chr6_aligned.map | Выравнивает риды на референс и записывает результат в chr6_aligned.map. Убрала параметр --no-spliced-alignment, потому что при секвенировании транскриптома разные части одной мРНК могут картироваться на экзоны, разделенные интронами. |
Вывод hiset2 выглядит следующим образом:
52939 reads; of these:
52939 (100.00%) were unpaired; of these:
8949 (16.90%) aligned 0 times
43990 (83.10%) aligned exactly 1 time
0 (0.00%) aligned >1 times
83.10% overall alignment rate
Из 52939 ридов, содержащихся в файле chr6_cleaned.1.fastq 83.10%, то есть 43990 ридов были откартированы ровно 1 раз на референсную последовательность хромосомы; 16.90% (8949 рида) не откартировались совсем, а больше одного раза ни один рид не откартировался.
3. Анализ выравнивания
| samtools view -bS chr6_aligned.map > chr6_aligned.bam | Переводит выравнивания из формата .map в формат .bam, результат - в файле chr6_aligned.bam |
| samtools sort -T ./tmp/sorted.tmp -o chr6_aligned_sorted.bam -O bam chr6_aligned.bam | Сортирует выравнивания по координате начала чтения в референсе. Результат - в файле chr6_aligned_sorted.bam |
| samtools index chr6_aligned_sorted.bam | Индексирует отсортированный .bam файл |
4. Подсчет чтений
| htseq-count -f bam -s no -i gene_id -m intersection-nonempty chr6_aligned_sorted.bam gencode.v19.chr_patchl_scaff.annotation.gtf -o chr6_count> | Опция -f указывает формат входного файла (bam или map), -s указывает, были ли входные данные точно только с одной цепи (у нас нет), -i указывает, что считать feature, в нашем случае - ген, -m указвает, что делать, если рид выравнивается или перекрывается более чем с одним feature. Либо учитывать все feature, либо брать пересечение всех feature (intersection-strict), либо брать пересечение только не пустых feature (intersection-nonempty). |
5. Анализ результатов
Выдача скрипта count.py:
__no_feature 171 (171 рид не лег в границы генов. Возможно, это риды из 3’UTR.)
__ambiguous 0 (ни один рид не откартировался на несколько генов одновременно)
__too_low_aQual 0 (ни один рид не ниже заданного порога, потому что порог не задан)
__not_aligned 8949 (не откартировалсось 8949 ридов)
__alignment_not_unique 0 (ни один рид не откартировался неоднозначно)
43819 ридов откартировались на ген ENSG00000156508.13, который кодирует фактор элонгации трансляции (eukaryotic translation elongation factor 1 alpha 1).