1. Подготовка чтений программой trimmomatic
Команды:
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 SRR4240360.fastq cutada.fastq ILLUMINACLIP:adapters.fasta:2:7:7 | Убирает из файла SRR4240360.fastq все адаптеры (адаптеры лежат в adapters.fasta) и записывает чтения без адаптеров в cutada.fastq. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 cutada.fastq cutada_2.fastq SLIDINGWINDOW:10:20 | Удаляет плохие буквы с концов чтений |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 cutada_2.fastq cleaned.fastq MINLEN:30 | Удаляет чтения длиной меньше 30 нуклеотидов |
| grep -c @ cutada.fastq | Смотрит, сколько чтений было до чистки: 8722538 |
| grep -c @ cleaned.fastq | Смотрит, сколько чтений стало: 8294738 |
2. velveth, velvetg
Команды:
| velveth velveth/ 29 -fastq -short cleaned.fastq | Готовит k-меры длины k=29, выходные файлы помещает в папку velveth/. |
| velvetg ./ | Сборка на основе k-меров |
N50=67095
| Контиг | Длина | Покрытие |
| 1 самый длинный | 94956 | 43.61 |
| 2 самый длинный | 70305 | 49.28 |
| 3 самый длинный | 70300 | 41.95 |
| Аномально маленькое покрытие | 4 | 1.5 (а среднее: 39,81) |
| Аномально большое покрытие | 1 | 148191 |
3. Анализ
С каждым контигом получилось очень много выравниваний, поэтому координаты участка хромосомы и соответствующие координаты контига и характеристики выравниваний - прямо в таблице из бласта.
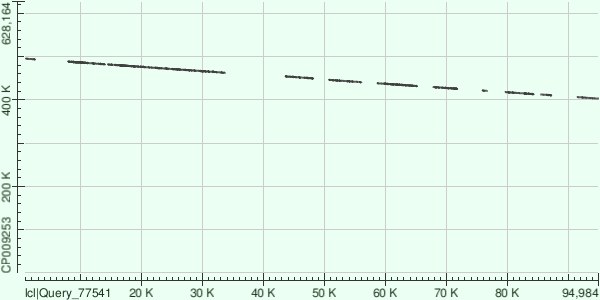
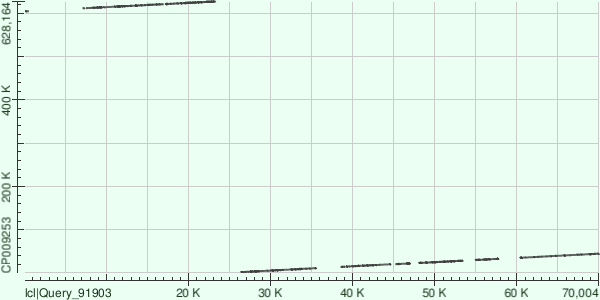
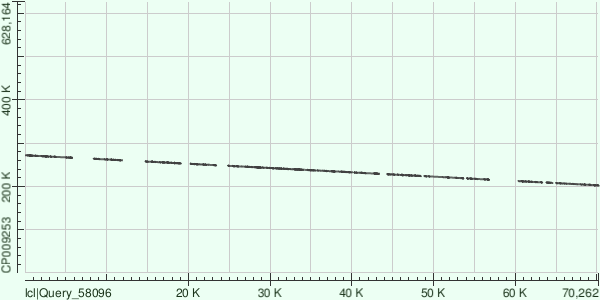
| Контиг длины 94956 | Контиг длины 70305 | Контиг длины 70300 |
| Получилось 20 выравниваний. Контиг ложится на обратную цепь хромосомы (что видно по наклону линии на матрице). Между выравниваниями довольно большие промежутки. | 13 выравниваний, контиг ложится на прямую цепь. Хромосома - кольцевая, контиг захватил место начала ее картирования (оно же начало репликации, наверное), и поэтому такой большой разрыв. | 14 выравниваний, контиг ложится на обратную цепь. |
| Таблица | Таблица | Таблица |
 |
 |
 |