Практикум 11.Ресеквенирование. Поиск полиморфизмов у человека.
Часть 1
Был сделан контроль качества чтений с помощью программы FastQC.
Команда: fastqc chr13.fastq
Было получено изображение (Рис.1)
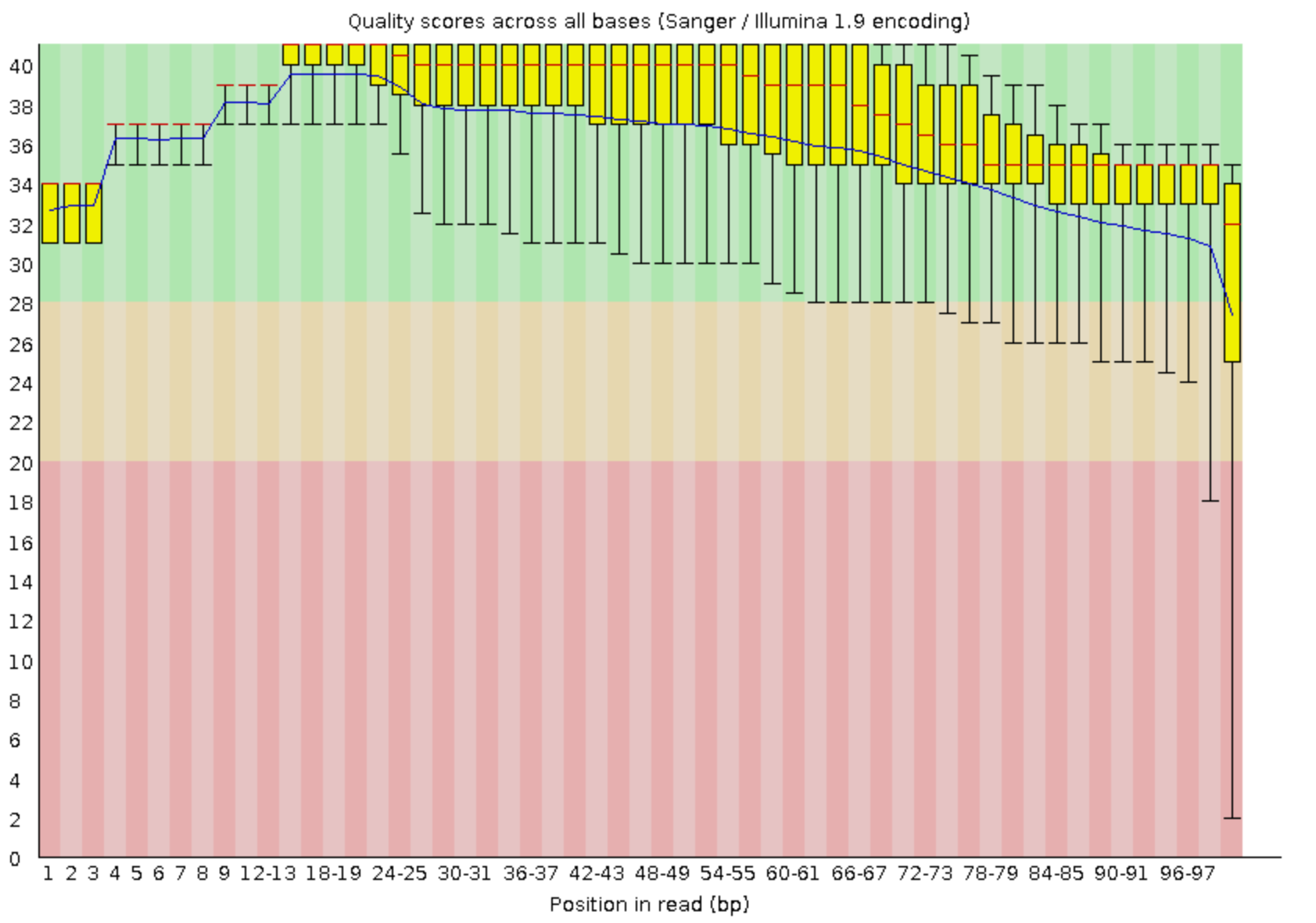
(Рис.1)
Затем была проведена очистка чтений при помощи программы Trimmonatic.
Команда: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq chr13_new.fastq TRAILING:20 MINLEN:50
После чистки качество прочтений представлено на Рис.2
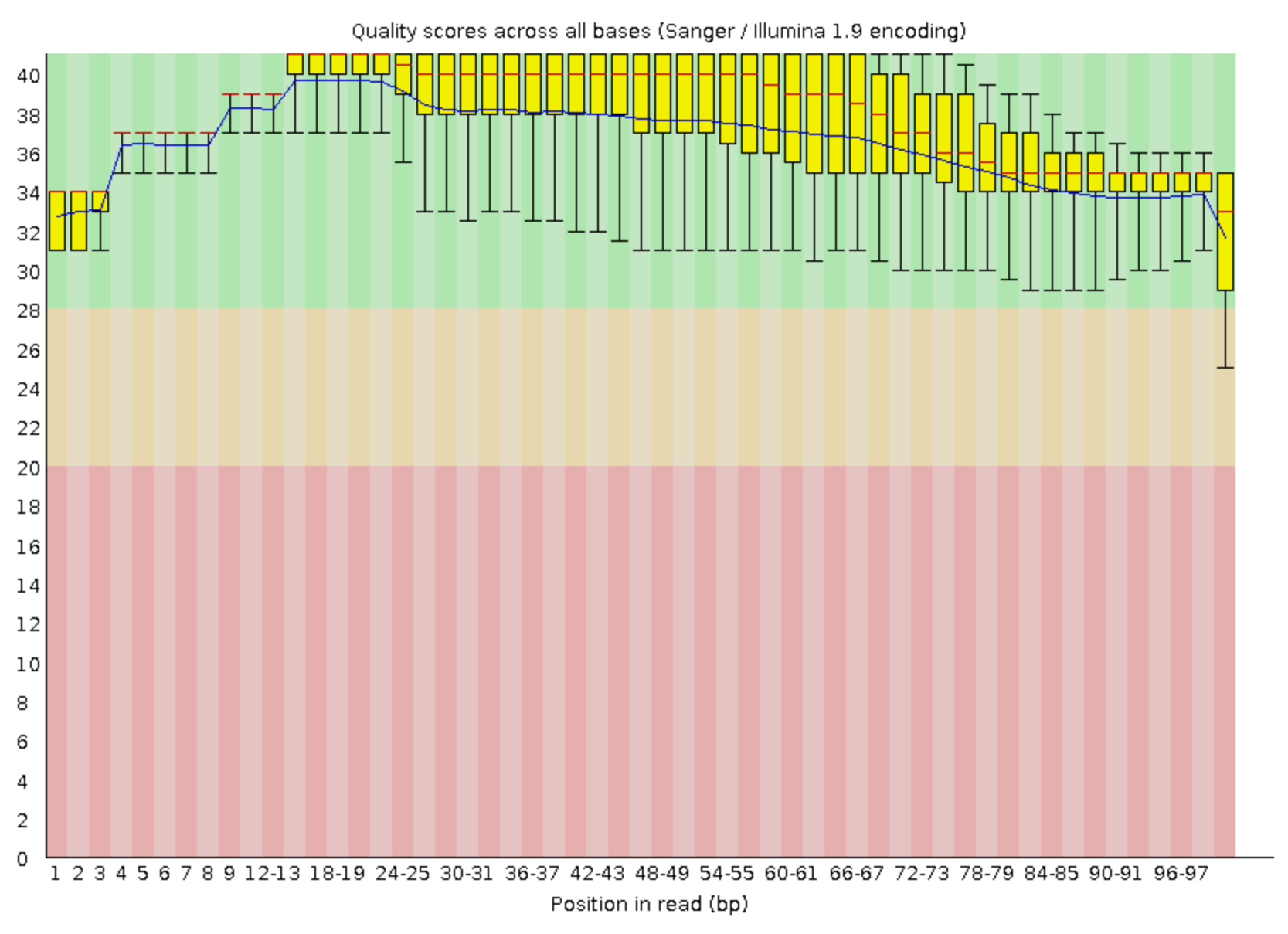
Рис.2
Изначально было 12155 прочтений с длинной 39-100, после чистки осталось 11933 с длинной 50-100.
Качество улушилось, теперь все позиции нуклеотидов по качеству попадают в зеленую область гистограммы,
что свидетельствует
о достоверности данных позиций. Также были обрезаны нуклеотиды с конца каждого прочтения,
с качеством ниже 20, что и наглядно отражено на гистограмме
(заметно короче стал последний столбик).
Часть 2
Картирование чтений
Далее была проиндексирована референская последовательность:
Команда:
hisat2-build chr13.fasta chr13_ind.fasta
Построено выравнивание прочтений и референса в формате .sam
Команда:
hisat2 -x chr13_ind.fasta -U chr13_new.fastq --no-spliced-alignment --no-softclip > align.sam
Анализ выравнивания
Выравнивание чтений с референсом было переведено в бинарный формат .bam.
Команда:
samtools view align.sam -b -o align.bam
Варавнивание чтений с референсом было отсортировано
Команда:
samtools sort -T /tmp/align.sorted -o align.sorted.bam align.bam
Затем проиндексировано
Команды:
samtools index align.sorted.bam
Чтобы понять какое количество чтений было картировано, был просмотрен вывод программы hisat2.
Она показала что 120 чтений
из 11933 не были картированы на референсную последовательность (это составило 1,01%)
Часть III: Анализ SNP
Поиск SNP и инделей.
Сначала был создан файл с полиморфизмами (формат .bcf)
Команда:
samtools mpileup -uf chr13.fasta align.sorted.bam -o align.bcf
И файл уже со списком отличий между референсом и чтениями (в формате .vcf)
Команда:
bcftools call -cv align.bcf -o align.vcf
Таблица полиморфизмов
| Координата |
Тип полиморфизма |
Референс |
Чтения |
Глубина покрытия |
Качество чтения |
| 20006620 |
Замена |
С |
Т |
19 |
221.999 |
| 25511130 |
Вставка |
Т |
ТСА |
8 |
196.47 |
| 25527657 |
Делеция |
GAAAA |
GAAA |
44 |
73.4665 |
Анотация SNP
С помощью программы annovar были проаннотированы полученные snp.
Предварительно из файла align.vcf были удалены индели
Всего было найдено 15 инделей.
При помощи convert2annovar.pl был создан файл с которым будет работать Annovar (команда:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 align.vcf > align.avinput)
Snp имеющие rs
Далее из 163 оставшихся snp 24 имеют rs.
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl
-filter -out rs -build hg19 -dbtype snp138
align.avinput /nfs/srv/databases/annovar/humandb/
Далее SNP были проаннотированы при помощи 5 баз данных.
1) Refgene
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl
-out refgene -build hg19 align.avinput /nfs/srv/databases/annovar/humandb/
В итоге были получены 3 файла, в одном из которых находились SNP.
Всего было найдено 163 SNP. В таблице приведены виды SNP.

В экзонных областях было найдено 9 SNP ,
4 из которых привели к заменам аминокислот. Все найденные SNP попали в гнеы:TPTE2,
PHF11 ,
COL4A1
4 замены произшли в следующих позициях:
| Координата |
До |
После |
Качество чтения |
Глубина |
| 110818598 |
T |
G |
212.000 |
49 |
| 110827684 |
T |
A |
15.1417 |
27 |
| 110804809 |
G |
A |
166.009 |
26 |
| 110813709 |
G |
A |
221.009 |
46 |
2)Dbsnp
Были проаннатированы по базе данных dbsnp
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter
-out dbsnp -build hg19 -dbtype snp138 align.avinput /nfs/srv/databases/annovar/humandb/
На выходе были получены 3 файла. В файле .filtered остались 24 snp.
3) Gwas
Аннотация по базе данных Gwas
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl
-regionanno -build hg19 -out gwas -dbtype
gwasCatalog align.avinput /nfs/srv/databases/annovar/humandb/
В итоге был получен файл, в котором остались 3 snp,имеющие клиническое значение.
| Координата |
Клиническое значение |
До |
После |
Качество чтения |
Глубина |
| 25533831 |
Полиморфизм связанный с ожирением |
A |
C |
225.009 |
78 |
| 50080847 |
Сердечная гипертрофия |
A |
G |
221.999 |
45 |
| 110818598 |
Жесткость артерий |
T |
G |
212.009 |
49 |
Два из трех (полиморфизм связанный с сердечной гипертрофией и жесткостью артерий)
находятся в экзонных областях, что может проявится на структуре белков, а значит и повлиять на здоровье человека.
4) Clinvar
База данных Clinvar
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter
-dbtype clinvar_20150629 -buildver hg19
align.avinput -outfile clinvar /nfs/srv/databases/annovar/humandb/
При этом не было найдено ни одного snp имеющего какое-либо клиническое значение.
5) 1000 Genomes:
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype
1000g2014oct_all -buildver hg19 -out 1000genoms
align.avinput /nfs/srv/databases/annovar/humandb/
В файле .filtered осталось 44 snp. Стоит отметить, что все snp, которые были найдены через dbsnp попали сюда.
Сводная таблица по базам данных