Введение
В ходе данного практикума было необходимо разобраться в работе алгоритма Blast. Также с помощью данной программы было необходимо найти гомологичные белки, а затем выровнять их в программе Jalview и проанализировать результаты, доказав гомологию данных последовательностей. Также было необходимо выровнять две белковых последовательности для получения карты локального сходства. Дополнительно необходимо было исследовать опции Blast и увидеть различия между поисками с разными параметрами.
Поиск гомологов ε-субъединицы АТР-синтазы Bacillus Subtilis с помощью алгоритмов Blast
Параметры Blast
Database: В данном окне можно выбрать базу данных, в которой алгоритм будет вести поиск. Для поиска доступны UniProt/PDB/Ref_Seq, а также нккоторые другие.
Organism: В данном окне можно указать организм, в котором будет вестись поиск. Можно указать несколько.
Algorithm: Выбор алгоритма поиска. Интересно, что часть алгоритмов позволяет осуществлять поиск по специфичным позициям в белках, по паттернам или доменам. Также доступен быстрый алгоритм поиска.
Max target sequences: Выбор количества показываемых находок. Находки сортируются по E-value и проценту гомологии.
Expect threshold: Установка ограничения на максимальное значение E-value.
Сам же параметр отражает математическое ожидание появления выравнивания с таким же или большим весом, как и осуществленное. Берется банк случайных последовательностей такого же размера, как и тот, по которому идет поиск гомологичных белков. Затем строится выравнивание исходной последовательности со случайными. Высчитывается нормализованный вес в байтах. Затем считается E-value по формуле E-value=mn·2^(-B), где -В — нормализованный вес в байтах. По сути, параметр E-value говорит нам о том, каков шанс получить выравнивание с таким же или большим весом для нашей последовательности и какой-то ОДНОЙ случайной той же длины. Параметр В показывает, что для получения выравнивания со случайной последовательностью с таким же или большим весом нужно провести 2^(B) выравниваний.
Word size: Параметр позволяет задать длину индексированных последовательностей. Blast разбивает все последовательности указанной базы данных на фрагменты заданной длины и сравнивает их с фрагментами исходной последовательности. Составляется таблица сходства; кусочки считаются похожими при определенном весе для каждого значения длины. Затем составляется таблица находок. Будут выравниваться те последовательности, имеющие 2 или более сходных кусков, которые по координатам будут совпадать с задаваемой последовательностью. Потом выравнивание осуществляется в две стороны от найденных кусочков.
Matrix: Позволяет выбрать матрицу аминокислотного сходства для подсчета счета выравнивания. Классическая — BLOSUM62.
Gap Costs: Выбор величины шрафа за гэп и за удлинение гэпов.
Compositional adjustments: Позволяет выбрать метод "борьбы" с участками малой сложности, которые могут приводить к неправильным результатам.
Поиск гомологичных белков
Был выполнен поиск гомологичных белков для ε-субъединицы АТР-синтазы сенной палочки. Был изменен только 1 стандартный параметр Blast: на экран выводились 20000 результатов поиска.

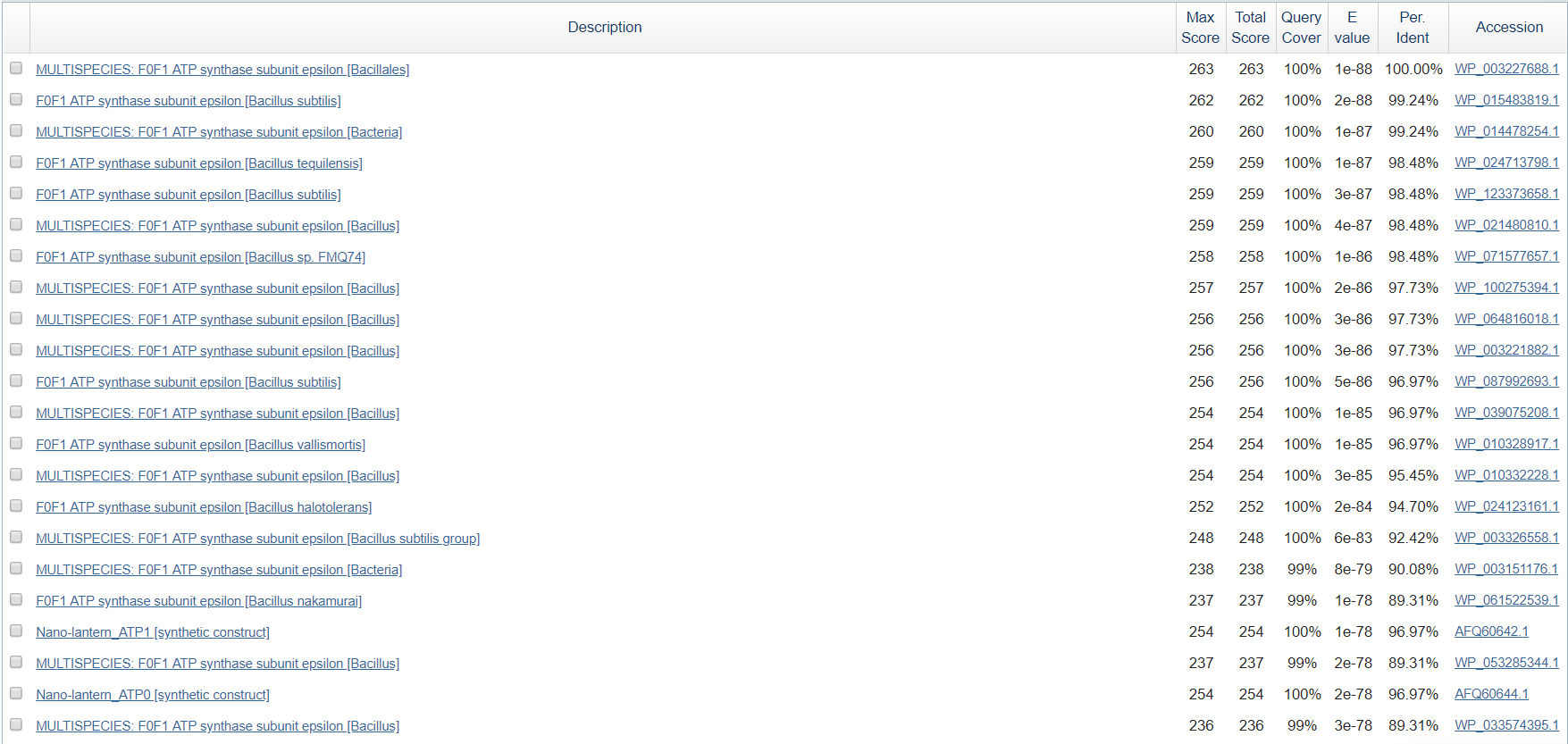
На картинке сверху показана цветовая схема для счета выравниваний последовательностей с наименьшим E-value. Как видно на картинке, последовательности имеют очень высокую степень гомологии и большинство из них незначительно отличаются по длине на С-конце.
Оказалось, что процент идентичности данной субъединицы очень высок среди части представителей рода Bacillus. Также высокий процент идентичности наблюдается при выравнивании с синтетическими конструкциями ε-субъединицы.
Выравнивание полученных белков
В результатах работы Blast были найдены предположительно гомологичные белки, их последовательности были скачены в формате fasta, а затем было построено множественное выравнивание в JalView алгоритмом с параметрами по умолчанию. Было получено следующее изображение (покраска аминокислот со 100%-ной консервативностью):

Данные последовательности принадлежат организмам из родов Bacillus (10 верхних), Anoxybacillus (11), Parageobacillus (12) и Geobacillus (13). Данные последовательности показывают высокую степень идентичности и очевидно гомологичны по следующим пунктам:
- С 31 по 42 позицию (12 аминокислот) находятся 11 консервативных остатков. На 33 позиции — очень близкие глутаминовая и аспарагиновая кислота.
- Второй такой участок расположен с 66 по 88 позицию (23 аминокислоты). На 66, 69, 73, 81 позициях стоят аминокислоты с очень похожими свойствами, а 16 позиций консервативны.
Карта локального сходства белков
В ходе данного задания алгоритмами Blast было составлено локальное выравнивание данных белков: D3AZP5_POLPP (Dihydro-6-hydroxymethylpterin pyrophosphokinase) и E6SLJ6_THEM7 (7,8-dihydroneopterin aldolase). Результаты выравнивания оказались следующими:
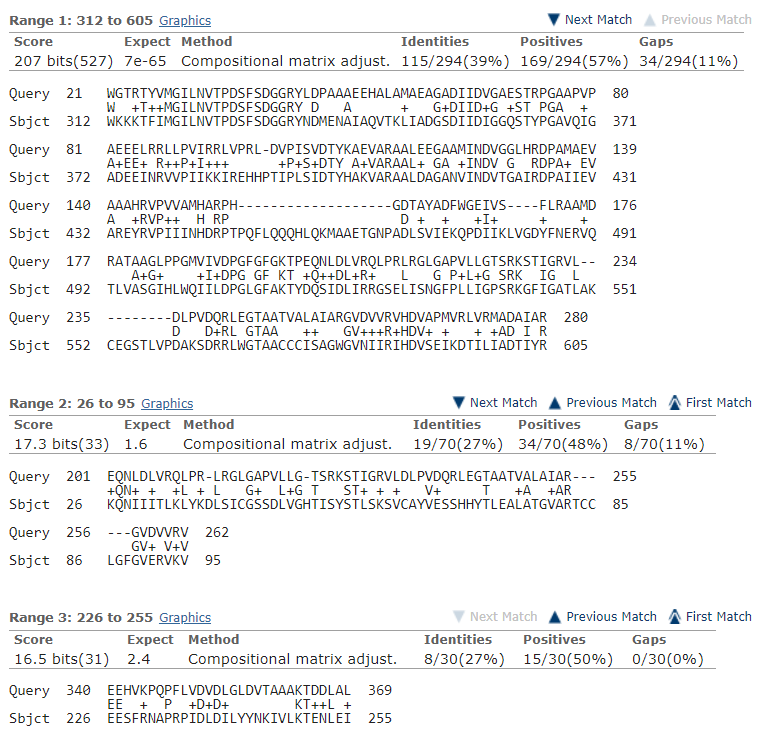
Blast выравнивает данные последовательности в трех местах, но так как для последних двух выравниваний очень велико E-value и очень мал счет выравнивания я не использую эти данные для дальнейшего анализа. Первое же выравнивание дает 57% похожих аминокислот, что достаточно много для негомологичных белков разных организмов.
Затем была скачана и проанализирована построенная карта локального сходства. Две короткие полосы в нижнем правом углу отражают результаты 2 и 3 выравниваний, поэтому не рассматриваются.
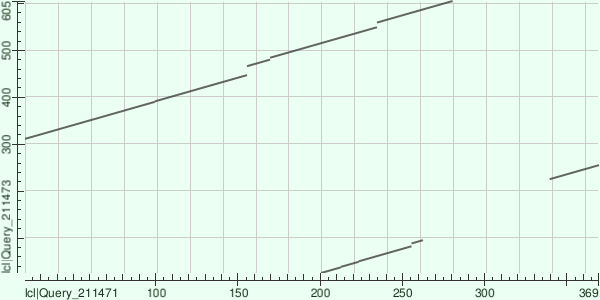
Из данной карты и результатов выравнивания можно сделать следующие выводы:
1) Так как последовательности имеют разную длину, у них не выравнивается начальные и конечные участки цепей.
2) Индели создают разрыв на графике локального сходства.
3) Данные белки имеют очень похожие участки, что и сознает высокий процент похожести и идентичности.
4) Первый белок выровнялся почти сначала и не до конца (график близок к 0 по Х, но вместе с тем не доходит до края шкалы), а второй наоборот в конце цепи (график далеко от нуля по У, но достигает пиковой точки по этой шкале).
В последовательности E6SLJ6_THEM7 происходит 3 достаточно крупных делеции (выпадают 19, 4 и 10 аминокислот). Вместе с тем данные последовательности несут много аминокислот с похожими свойствами (поля с плюсами), да и совпадает у них почти 40% остатков. Для них можно было бы предположить гомологию, но эти сходства могут оказаться случайными, так как данные белки катализируют разные реакции и данное сходство последовательностей может быть конвергентным.
Изменение параметров Blast
Небелковые последовательности
Для поиска гомологичных небелковых последовательностей Blast изменяет свои стандартные опции. Доступны другие базы данных (забавно, что можно искать гомологичные последовательности в мышином геноме и транскриптоме). Теперь можно оптимизировать программу поиска под свои цели: Blast умеет искать очень похожие, не очень похожие и последовательности, схожие в некоторых местах. Изменяется и диапазон Word size: доступен выбор блоков от 16 до 256 аминокислот. Изменяются и параментры счета выравнивания: их всего 2 — очки ха метч и штрафы за мисметч. Также можно устанавливаливать линейные и аффинные штрафы за гэпы. Доступен новый раздел "Filters and Masking".
Для нуклеотидных последовательностей я решила начать с параметра с Word size. Для гена рибосомальной 5s RNA из человека при значении этого параметра в 256 букв и при поиске в базе данных others Blast не находит ни одного значительного соответствия. Для длины в 28 нуклеотидов находится 11657 последовательностей. Для длины в 64 нуклеотида — 1337 последовательностей, причем самой гомологичной оказывается последовательность слона. Вариация этого параметра позволяет находить белки разной степени гомологии, а также может делать поиск более точным.
Также следует отметить, что минимальное значение E-value не меняется при изменении длины поисковых слов и составляет 5e^(-55), причем это значение одинаково для примерно первого десятка последовательностей. Возможно, это связано с математическими или вычислительными ограничениями алгоритма.
Также я посмотрела изменение числа находимых последовательностей при уменьшении E-value для длины слова 32. Для максимального значения в 10 алгоритм выдает 9861 последовательностей, для значений 1, 0.1, 0.001 число находок уменьшается. Для значения в 0.00001 алгоритм находит 9858 последовательностей, для 0.0000001 — 9851. Отсюда видно, что для нуклеотидных последовательностей изменения E-value должно быть очень большим, чтобы число находок поменялось кардинально.
Белковые последовательности
Я продолжаю использовать последовательность ε-субъединицы АТР-синтазы сенной палочки.
Сначала я посмотрю на различные минимальные E-value для разных параметров Word size. Word size 2 дает минимальное значение, равное 1e^(-88), Word size 3 — 1e^(-88), a Wordsize 6 такое же, как и первые 2. Но вместе с тем это значение не сохраняется на дальнейшие находки ни в одном случае. Возможно, это верхняя граница вычисления для метода.
Затем я исследовала зависимость E-value при Wors Size 6 от матрицы аминокислотных соответсвий. BLOSUM62 дает значение 1e^(-88), BLOSUM90 — 2e^(-90), PAM30 — 3e^(-81) и 16690 находок, PAM250 — 4e^(-73) и более 200 находок. Для последней матрицы быстро падает счет выравниваний:
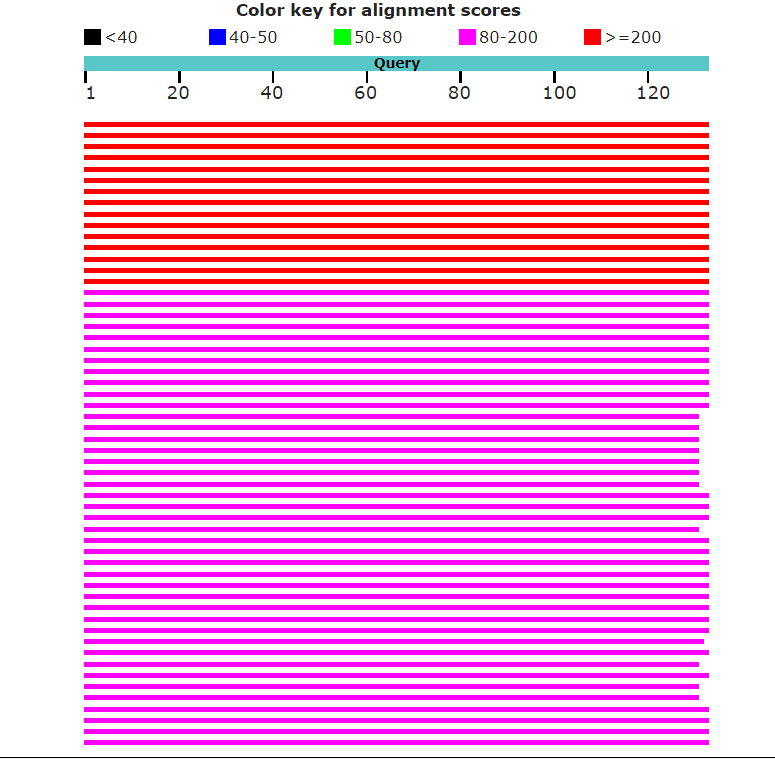
Изменение матрицы аминокислотных соответствий (ее подбор под каждый конкретный белок) позволит улучшить точность поиска.
Затем я попробовала изменить алгоритмы поиска.
Алгоритм PSI-Blast со стандартными значениями выдает 500 последовательностей, причем E-value min равно 1e^(-88). Этот тип алгоритма опирается на выравнивания по отдельным элементам.
Алгоритм DELTA-Blast со стандартными значениями выдает 500 последовательностей с E-value min равным 1e^(-62). Этот алгоритм выравнивает последовательности исходя из их доменной организации.
Изменения данного параметра помогут сделать поиск более специализировнным, так как белки с различными белковыми междоменными структурами могут быть гомологичны (домены консервативны, междоменные части вариабельны).