В ходе выполнения практикума я работала с 9 хромосомой человеков. В ходе выполнения практикума использовались следующие команды (указаны в хронологическом порядке):
| hisat2-build | Команда используется для индексирования референсной последовательности |
| fastqc | Команда используется для оценки качества ридов. На выходе получаем архив с html-страничкой. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9_1.fastq 91-50.trimmed.fastq TRAILING:20 MINLEN:50 | Команда используется для улучшения качества ридов (удаляет нуклеотиды с качеством менее 20 и риды общей длиной менее 50). |
| PATH=$PATH:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Команда указывает путь к файлам программы hisat2-2.0.5 |
| hisat2 -x chr9 -U 91-50.trimmed.fastq -S reads.sam --no-spliced-alignment --no-softclip | Команда накладывает риды на референсный геном и записывает их в выходной файл reads.sam. --no-spliced-alignment указывает, что рид должен лечь непрерывно, --no-softclip не позволяет наложиться какой-то части рида, игнорируя то, что на концах. |
| samtools view -b -o reads.bam reads.sam | Команда переводит файл с картированными ридами из формата .sam в формат .bam. |
| samtools sort reads.bam reads_sorted | Команда сортирует риды в соответствии с из порядком в индексированном геноме. |
| samtools index reads_sorted.bam | Команда индексирует риды |
| samtools mpileup -uf chr9.fasta -o polymorphisms.bcf reads_sorted.bam | Команда создает файл с полиморфизмами |
| bcftools call -cv polymorphisms.bcf -o polymorphisms.vcf | Команда меняет формат файла |
| vcftools --vcf polymorphisms.vcf --remove-indels --recode --out only_SNP | Команда вырезает индели из файла с полиморфизмами |
| convert2annovar.pl -format vcf4 only_SNP.recode.vcf > poly_SNP.avinput | Команда меняет формат файла |
| annotate_variation.pl -filter -out snp138_annotated -build hg19 -dbtype snp138 only_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по базе snp138. |
| annotate_variation.pl -filter -out ref_Gene_annotated -build hg19 -dbtype refGene only_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по базе ref_Gene. |
| annotate_variation.pl -filter -out 1000_genomes__annotated -build hg19 -dbtype 1000g2014oct_all only_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по базе 1000 геномов. |
| annotate_variation.pl -filter -out GWAS__annotated -build hg19 -dbtype gwasCatalog only_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по базе GWAS. |
| annotate_variation.pl -filter -out Clinvar_annotated -build hg19 -dbtype clinvar_20150629 only_SNP.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по базе Clinvar. |
Триммирование ридов
Качество ридов до триммирования
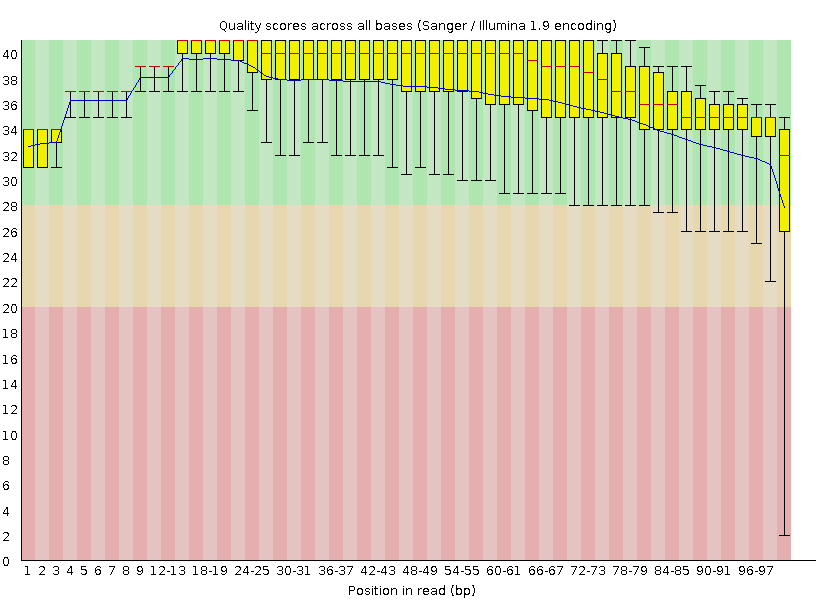
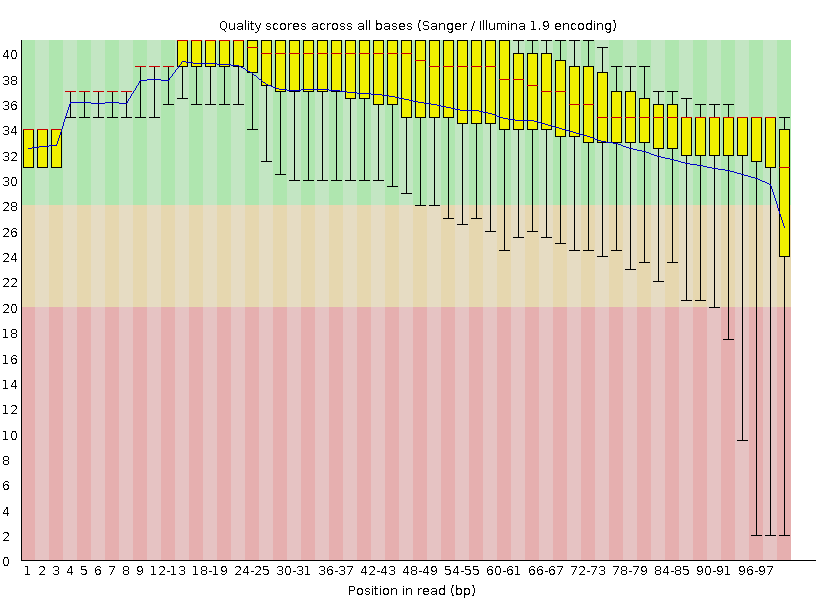
После триммирования общее качество ридов растет: из первого файла остается 10536 (из 10701) рида, а из второго — 2141 (из 2206). Как видно, число удаленных ридов не превышает примерно 2% от общего их числа.
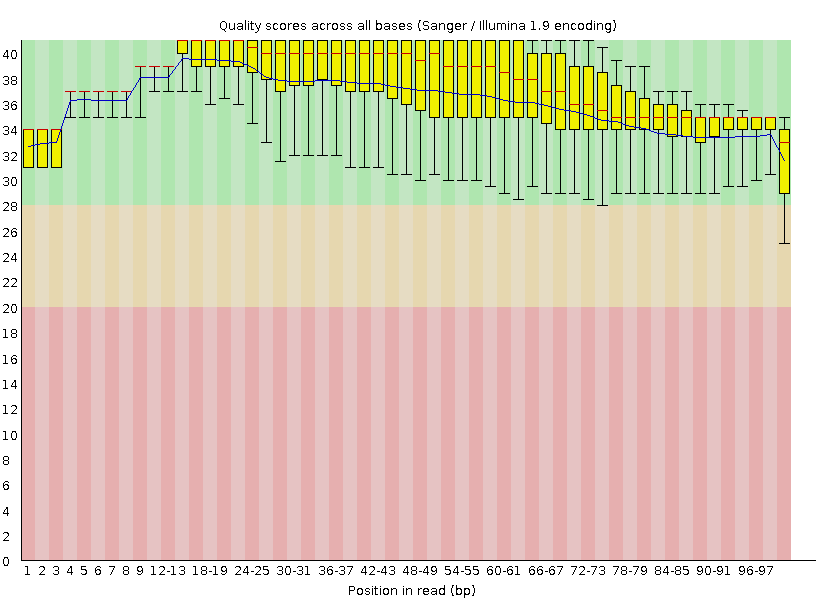
После триммирования количество ридов падает незначительно. Также растет их качество, но в моем случае этот рост не огромен, но так как он все же есть в условиях удаления малого числа ридов я считаю проведение данной процедуры оправданным.
Картирование ридов на геном
10536 reads; of these:
10536 (100.00%) were unpaired; of these:
73 (0.69%) aligned 0 times
10461 (99.29%) aligned exactly 1 time
2 (0.02%) aligned >1 times
99.31% overall alignment rate
Выше представлена выходный данные по работе картировщика. Как мы видим, больше 99% ридов ложатся на геном только один раз, 2 рида — 2 и более раз, а 73 рида не ложатся на референсный геном. В целом, можно отметить высокое качество картирования ридов.
Поиск SNP и инделей
Описание 3 полиморфизмов, которые были найдены в картированных ридах.
| Номер | Координата | Тип | Референс | Риды | Глубина покрытия | Качество чтения |
| 1 | 136133380 | INDEL | CGGGG | CG | - | 214.475 |
| 2 | 136136770 | SNP | A | C | 16 | 222.639 |
| 3 | 136137657 | SNP | C | T | 26 | 96.0077 |
Суммарное количество найденных полиморфизмов равно 111, из них инделей ровно 5 штук.
Среднее качество 81,86070586 Медианное качество 31,5765 Средняя глубина 12,16981132 Медианная глубина 3
В ходе анализа полиморфизмов стало понятно, что риды картируются на геном неравномерно, половина из них накладывается на геном 3 и менее раз (видно по медианам).
Ниже описаны различные типы SNP в базе данных RefSeq.
| exonic | 15 |
| splicing | 0 |
| intronic | 78 |
| ncRNA | 0 |
| UTR5 | 0 |
| UTR3 | 8 |
| upstream | 0 |
| dowmstream | 2 |
| intergenic | 3 |
SNP попадают в 5 генов: ABO, GLIS3, IL33, JAK2, SLC1A1. Для трех замен указан результат замены (2 синонимичные замены и одна несинонимичная):
synonymous SNV JAK2:NM_004972:exon19 nonsynonymous SNV JAK2:NM_004972:exon24 synonymous SNV IL33:NM_001199641:exon3
RS имеют 96 из 106 SNP. Информация взята из выхода программы аннотации по snp168.
Средняя частота SNP по базе 1000 genomes составила 42%.
3 замены аннотированы по Clinvar, но никакой дополнительной информации о них не дано.
8 SNP аннотированы в GWAS:
Crohn's disease: chr9 4985879 Endometriosis: chr9 6253571 Tumor biomarkers,Coagulation factor levels: chr9 136131188 mean corpuscular hemoglobin concentration: chr9 136131322 End-stage coagulation: chr9 136131415 Malaria: chr9 136132754 Activated partial thromboplastin time,D-dimer levels,Venous thromboembolism: chr9 136137065 Coagulation factor levels: chr9 136137106
3 SNP ассоциированы с болезнями: малярия, болезнь Крона и эндометриоз; остальные же сопряжены фенотипическими признаками, связанными с состоянием крови: факторы коагуляции и их уровень, активация тромбопластина. Это может говорить о том, что на 9 хромосоме человека закодировано множество генов, ответственных за механизмы свертывания крови.