В ходе выполнения практикума я работала с 9 хромосомой человеков. В ходе выполнения практикума использовались следующие команды (указаны в хронологическом порядке):
| hisat2-build | Команда используется для индексирования референсной последовательности. Индексирование было проведено в предыдущем практикуме. |
| fastqc chr9.1.fastq | Команда используется для оценки качества ридов. На выходе получаем архив с html-страничкой. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9.1.fastq trimmed.fastq TRAILING:20 MINLEN:50 | Команда используется для улучшения качества ридов (удаляет нуклеотиды с качеством менее 20 и риды общей длиной менее 50). |
| hisat2 -x chr9 -U trimmed.fastq -S reads.sam --no-softclip | Команда накладывает риды на референсный геном и записывает их в выходной файл reads.sam. --no-spliced-alignment указывает, что рид должен лечь непрерывно, поэтому здесь мы его убираем, так как прочитаться могли части экзонов в зрелой мРНК, которые в геноме разделены интроном, --no-softclip не позволяет наложиться какой-то части рида, игнорируя то, что на конце. |
| samtools view -b -o reads.bam reads.sam | Команда переводит файл с картированными ридами из формата .sam в формат .bam. |
| samtools sort reads.bam reads_sorted | Команда сортирует риды в соответствии с из порядком в индексированном геноме. |
| samtools index reads_sorted.bam | Команда индексирует риды. |
| htseq-count -f bam mapped_reads.bam -i gene_id -s no gencode.v19.chr_patch_hapl_scaff.annotation.gtf > Counts | Команда считает число ридов, которые попали на каждый из генов. |
| grep -wv 0 Counts > results.txt | Записывает в текстовый файл строчки, не заканчивающиеся на 0 (т.е. те гены, на которые произошло наложение). |
Оценка качества ридов
Качество ридов до триммирования
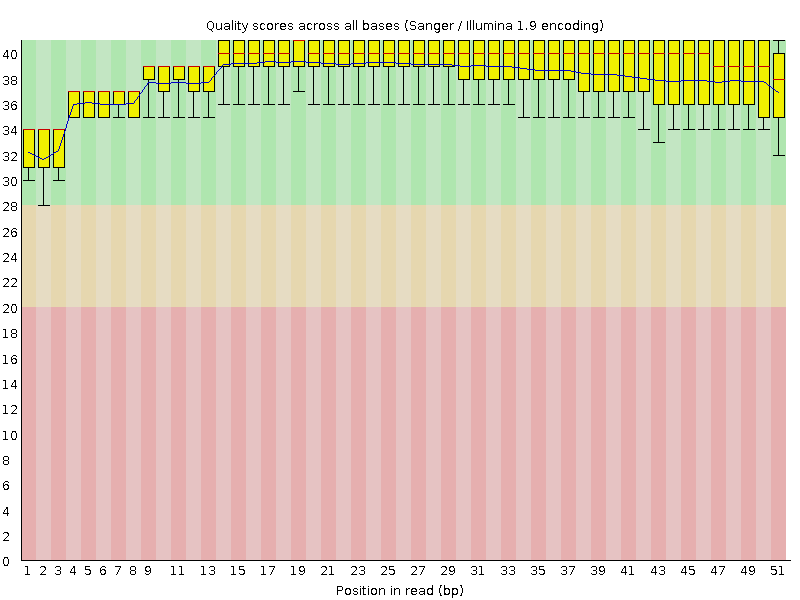
Процедура триммирования прошла вот так вот:
Input Reads: 19976 Surviving: 19267 (96,45%) Dropped: 709 (3,55%)
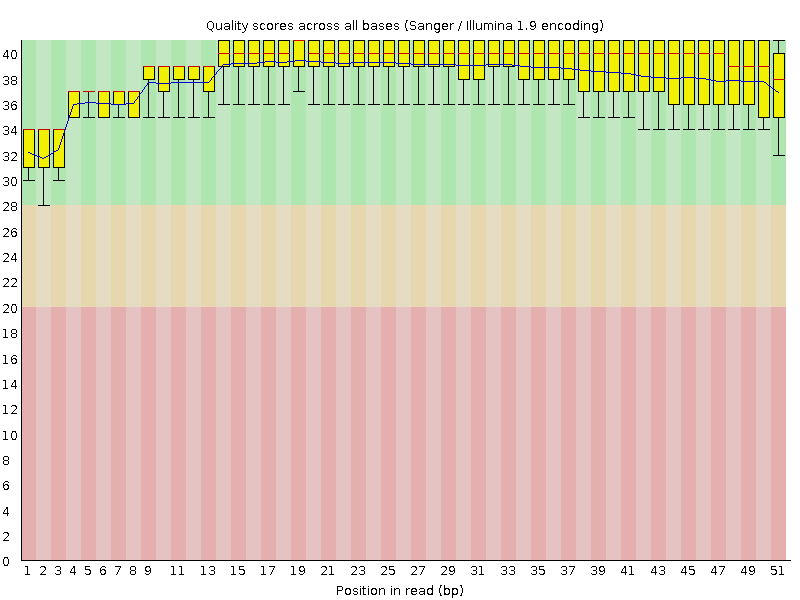
Визуально качество ридов растет, но очень слабо.
Картирование ридов
19267 reads; of these:
19267 (100.00%) were unpaired; of these:
113 (0.59%) aligned 0 times
19154 (99.41%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.41% overall alignment rate
113 ридов не накладываются на референсный геном, два и более раз не накладывается ни один рид.
Подсчет чтений
Опции htseq-count и где они обитают
Программа подсчитывает, сколько ридов ложатся на какой-то конкретный ген. Ниже описаны некторые из ее опций.
-i: GFF атрибут, который используется в качестве feature ID. -f: указание на формат bam или sam. -s: [yes, no, reverse] позволяет указать, относятся ли риды к прямой, обратной или обеим (через "no") цепям. -m: [union, intersection-strict, intersection-nonempty] определяет, какие именно наложения рида считать наложением на ген.
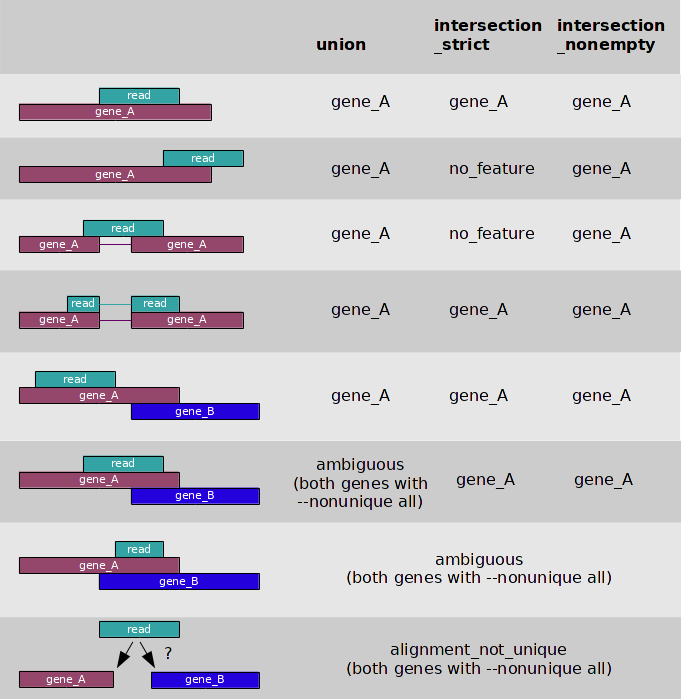
Анализ наложений ридов на гены
ENSG00000119335.12 18877 __no_feature 277 __not_aligned 113
Выше представлен результат работы grep, в котором видно, что все риды накладываются всего лишь на один единственный ген. Небольшое количество ридов на него не накладываюся. Это могли оказаться, например, поли-А концы созревшей мРНК, или же риды могли содержать часть промоторной области.
Интересно, что все риды ложатся на протоонкогенный ген, который кодирует белок, ингибирующий ацелирование гистонов в нуклеосомах. Предполагается, что он защищает лизины от ацилирования с помощью подавления HAT-индуцированной транскрипции. Обычно он локализуется в ЭПР, но был найден и в ядре, где предотвращал апоптоз клеток, который был индуцирован Т-киллерами. Для белка найдены несколько изоформ.