Определение таксономии для прочтенной последовательности
Для определения таксономии полученной в 6 практикуме консенснусной последовательности было запущено 2 алгоритма Blast: megablast и blastn. Результаты выдачи алгоритмов совпадали (ниже представлена выдача для blastn).
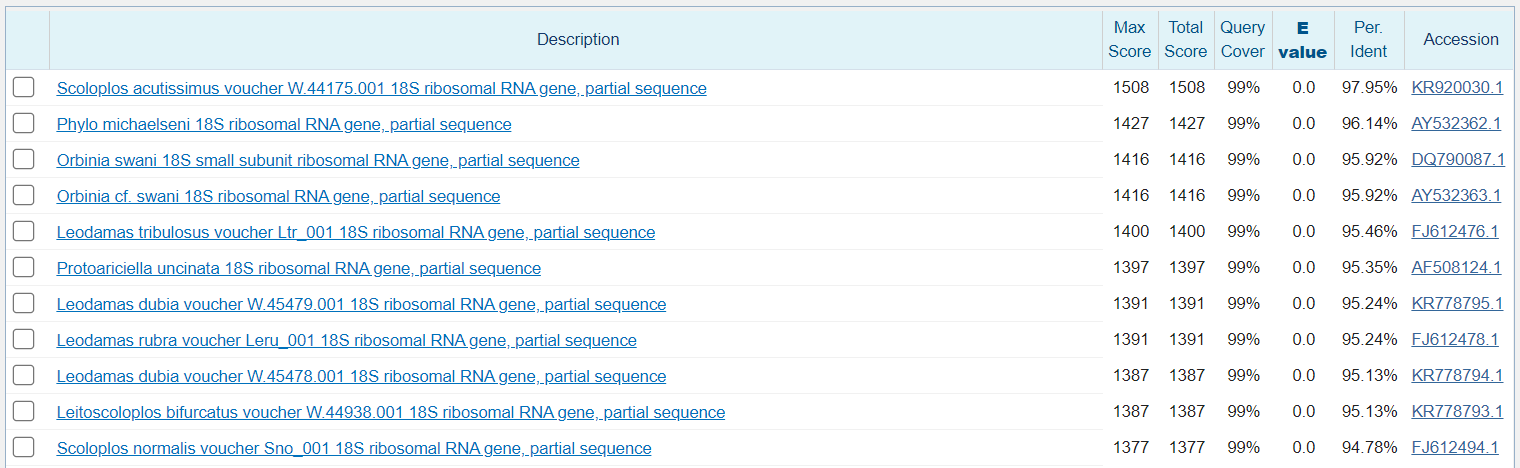
Низкий E-value, высокий процент покрытия и идентичности позволяет утверждать, что отсеквенирован был ген 16S RNA. Ниже показано лучшее полученное выравнивание.

Данное выравнивание, как и другие полученные, подтверждает то, что отсеквенирован был ген одной из рибосомальных РНК. Во-первых, почти 100% нуклеотидов совпадают на позициях ~100-600 в консенсусной последовательности, для которой характерно самое лучшее качество секвенирования и однозначное двойное прочтение каждого нуклеотида. Во-вторых, общее число несовпадений в выравнивании составляет 16 на 881 нуклеотид, процент ошибки — 1,8. Причем, ошибки могут быть связаны как и с ошибками в обоих секвенированиях, так и с ошибкой при интерпретации данных с секвенатора. В третьих, все 100 находок отсылают к генам 18S РНК, что позволяет однозначно установить секвенированный ген.
Данный ген не транслируется в белок, а как РНК-транскрипт входит в состав малой субъединицы рибосомы.
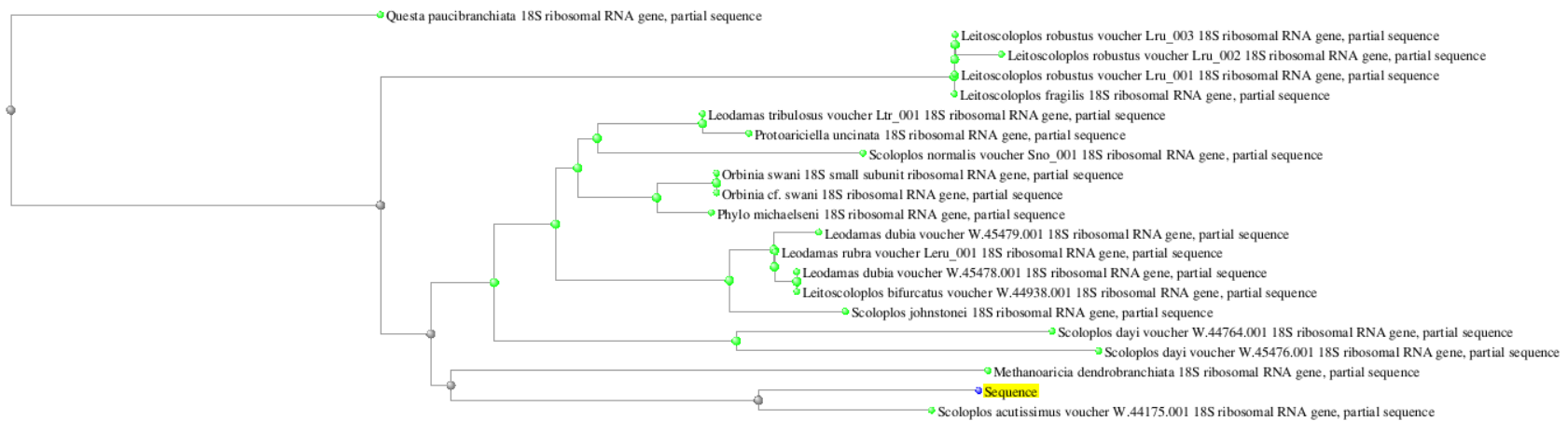
В дальнейшем с использованием тех же выдач алгоритма было установлено таксономическое положение организма, из которого была отсеквенирована последовательность. Я воспользовалась деревом для первых 20 последовательностей, который выдает алгоритм, а также гуглом. Самая дальняя находка имеет минимальный score и coverage, поэтому я не учитываю ее при определении таксономии. Однозначно можно установить, что эта последовательность получена из многощетинкового кольчатого червя инфракласса сколециды, семейства Оrbiniidae.
Википедия говорит об этом семействе вот так:
Морские придонные многощетинковые черви с удлиненной формой тела. Вид Berkeleyia hadala встречается на глубинах от 3086 до 6143 м. Как правило, тело разделено на широкую дорзо-вентрально сплющенную грудную область и заднюю брюшную более узкую и округленную в поперечном сечении и состоящую из сегментов, несущих дорзальные параподии.
Наибольшее число находок принадлежит роду Scoloplos, так что можно предположить, что это последовательность получена из вида этого рода или близкого к нему. Черви данного вида обитают в достаточно широкой географической зоне, в том числе и на Белом море (возможно, хроматограммы прямиком с ББС).
Сравнение результатов выдачи разных алгоритмов Blast
Запуск алгоритма для консенснусной последовательности
Megablast
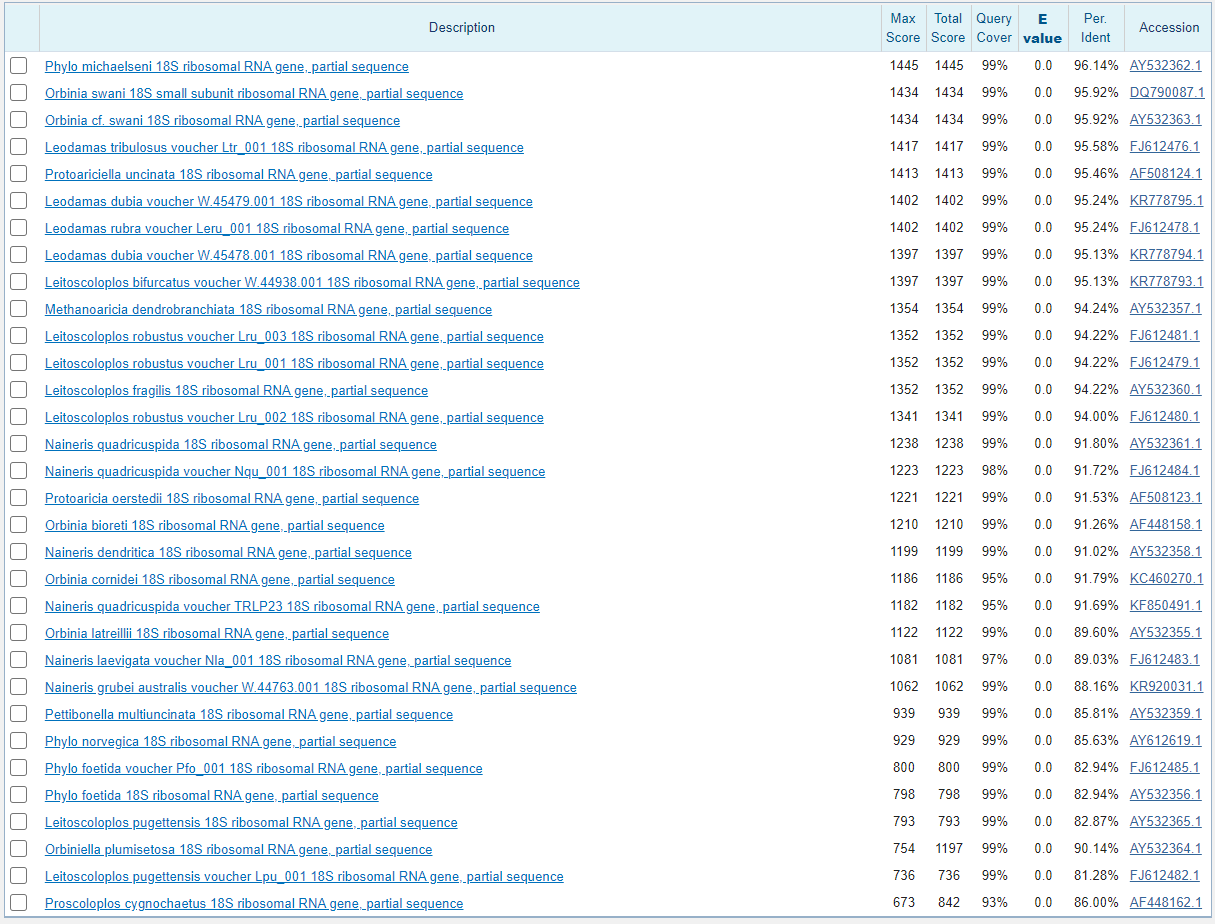
Тут, как и во всех дальнейших случаях, поиск осуществляется по семейству, исключая род, с выдачей в 100 находок. Для этого поиска я выбрала длину слова в 24 буквы.
Алгоритм выдает 32 находки с очень высоким E-value и процентом идентичности выше 80. Также в выдаче присутсвуют только гены 18S rRNA.
Discontiguous megablast
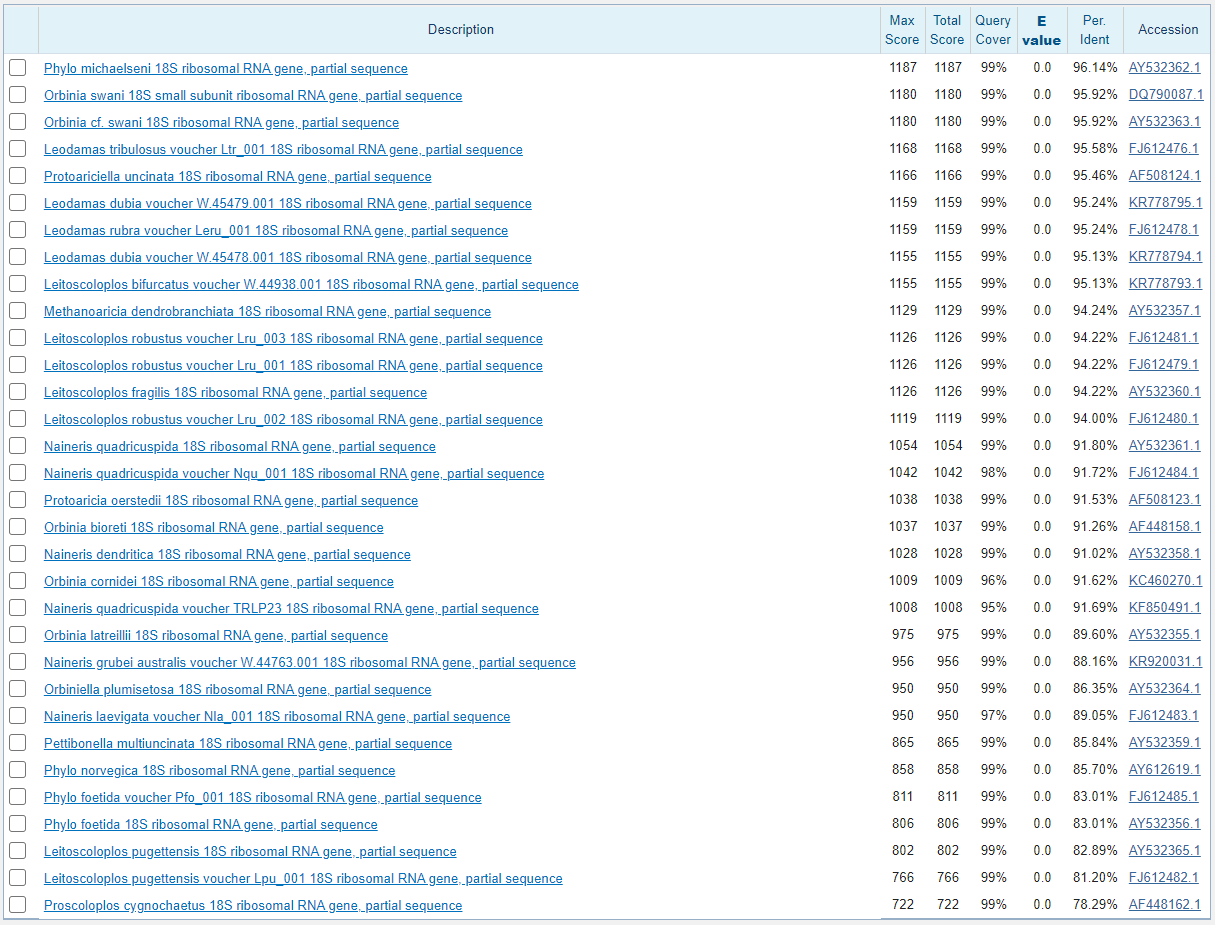
Поиск осуществлялся по длине слова 11 и штрафами за гепы в 2/2. Смена параметров поиска (в том числе и этих), не изменяла количество находок: их все равно остается 32. Результаты поиска немного отличаются скором для идентичных последовательностей, особенно это видно на Protoscoloplos cygnochaetus (последняя строка выдачи для обоих алгоритмов). Для megablast Total score и процент идентичности данной последовательности выше, чем для второго алгоритма, что позволяет сказать, что megablast осуществляет более эффективные выравнивания.
Чувствительный Blastn

Поиск осуществлялся по длине слова 7, чтобы снизить число находок с очень высоким E-value.
Поиск выдает 46 находок, из которых верхние 26 неплохо совпадают с находками других алгоритмов. Они имеют самый низкий E-value, процент идентичности более 89 и наивысший max score. Но вместе с тем алгоритм находит еще несколько последовательностей (8) с более высокими значениями E-value, но все еще являющимися гомологами консенснусной последовательности (тоже являются генами 18S rRNA). Остальные же находки имеют очень высокий E-value (больше 1) и вообще представляют собой абсолютно негомологичные последовательности (особенно цитохром оксидаза).
Blastn
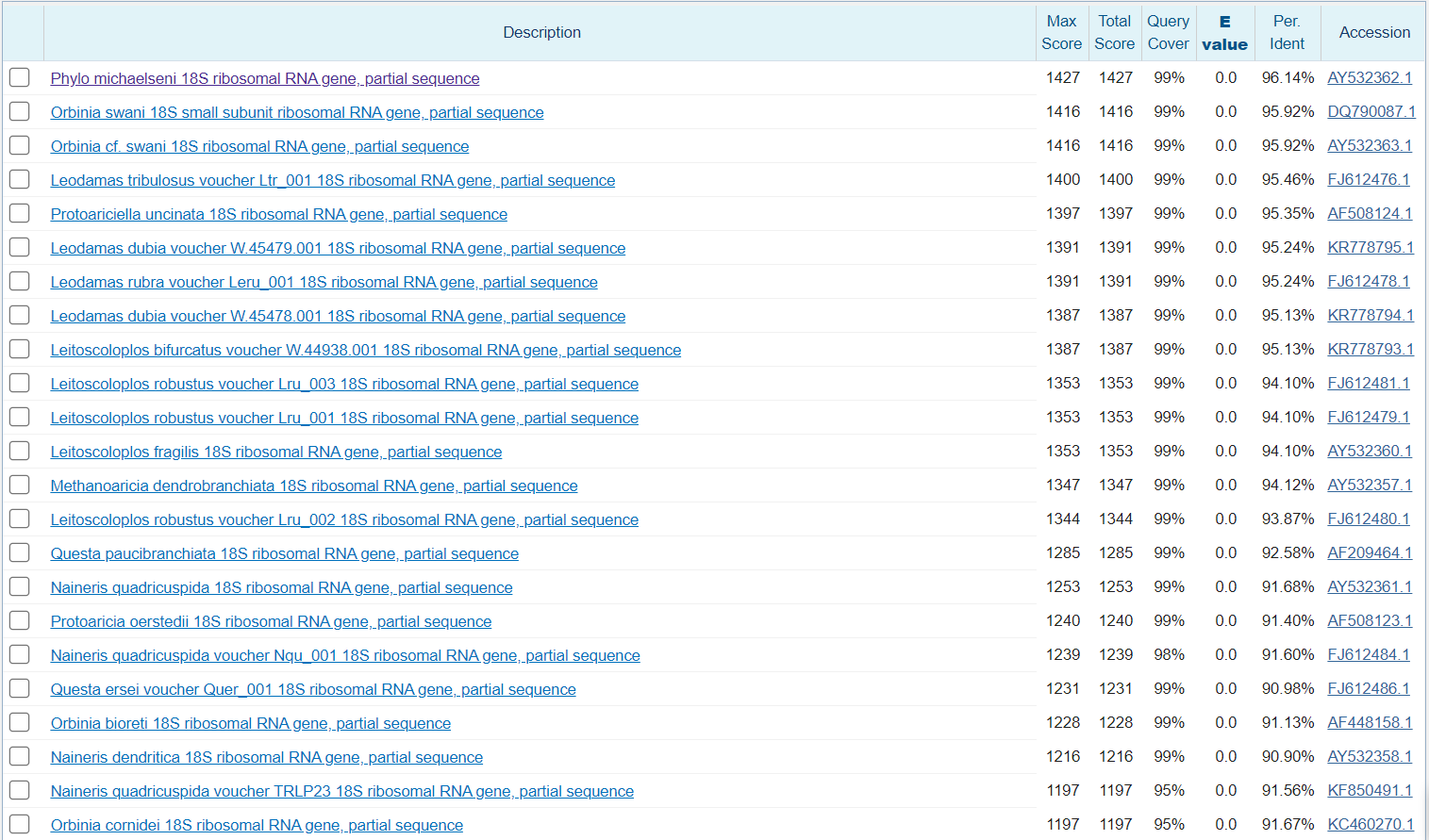
Поиск осуществлялся по длине слова 11.
Поиск выдает 99 находок с высоким E-value. Все из них относятся к генам рибосомальной 18S RNA. Многие находки совпадают с выдачей чувствительного blast и megablast.
Таким образом, алгоритм blastn является более гибким и позволяет находить большее число гомологов интересующей нас последовательности, в то время как более точные алгоритмы могут использоваться для более тонких целей. Например, в теории, ими можно искать ошибки в секвенированиях, если имеется большое число известных последовательностей (например, для человеков или арабидопсисов).
Запуск алгоритма для CDS
Megablast

Алгоритм здесь и в дальнейшем запускался для гена, кодирующего хеликазу (putative superfamily 2 helicase) среди организмов семейства Lipothrixviridae. Параметры запуска отличались от стандартных: использованы wordsize 32, existence/extension 2/2.
Алгорит находит всего 2 находки, причем среди организмов одного вида с высоким процентом идентичности и скором, что еще раз указывает на то, что алгоритм заточен только на поиск близкородственных последовательностей.
Discontiguous megablast
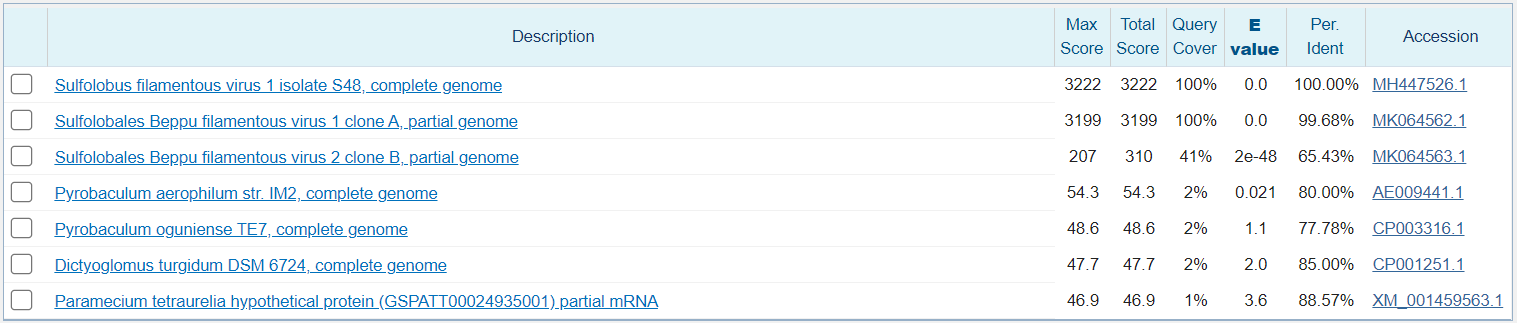
Был изменен 1 стандартный параметр: match/mismatch 5/-3. Алгоритм выдает всего 7 находок, но только 3 из них релеватны по E-value. Он находит еще один подвид данного вируса, но скор для данного организма оказывается в 10 раз ниже, чем для такого же подвида 1 типа, что может свидетельствовать о огромных генетических различиях между подвидами. Остальные результаты нерелевантны, так как имеют высокий E-value, причем удивительным образом там выскакивает даже инфузория, которая вряд ли имеет общие с вирусом последовательности.
Чувствительный Blastn
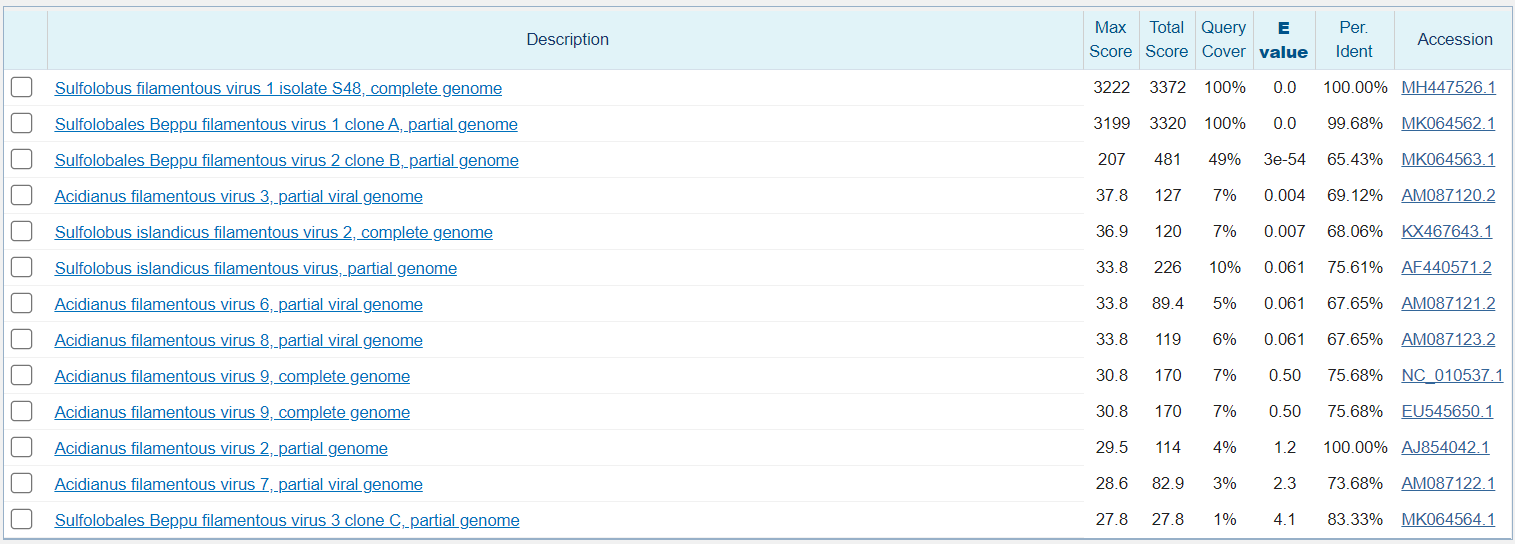
Измененные параметры: match/mismatch 4/-5, existence/extension 12/8.
Алгоритм находит всего 13 последовательностей, но релевантных среди них снова только 3. Остальные хоть и имеют высокий процент идентичности, но несут низкий score и E-value, но вместе с тем принадлежат тому же роду вирусов.
Blastn
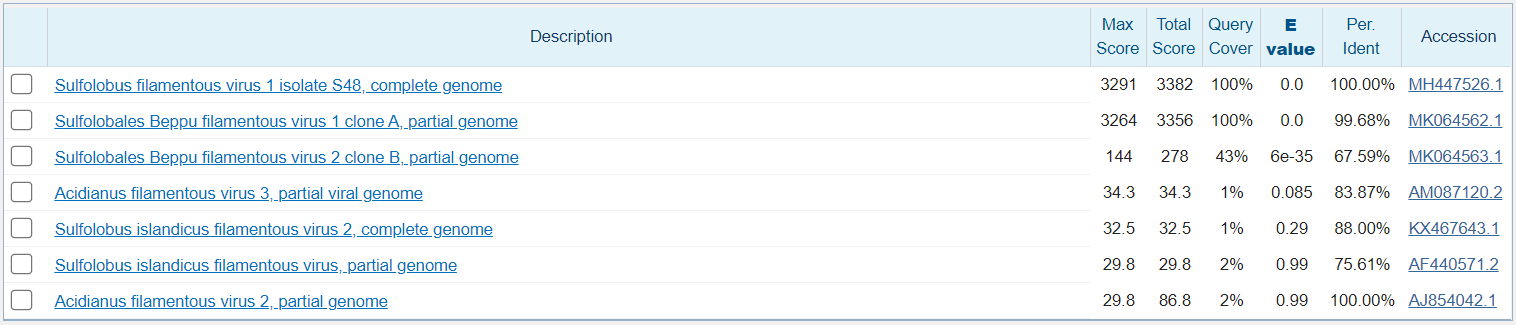
Измененные параметры: existance/extension: 2/4
Алгоритм находит 7 последовательностей, что странно, так как поиск должен был быть менее чувствительным. Релевантных находок все еще только 3, а все остальные — из видов близких родов.
А по итогу?
Blastn во всех случаях находит сильно больше последовательностей, но требует более критичного подхода к результатам поиска: иногда он выдает всякие странные вещи (например, роднит рибосомальную РНК и цитохром оксидазу), но вместе с тем несет больше информации. Megablast же сильно комфортней: выдaет только очень похожие штуки с высоким скором. Второй же алгоритм сочетает в себе достоинства и недостатки вышеописанных.
А теперь табличка без границ, которая написана в ночи.
| Алгоритм | Консенсус | CDS |
| Megablast | 22 | 2 |
| Discontiguous megablast | 22 | 7 |
| Blastn | 99 | 7 |
| Чувствительный blastn | 46 | 13 |
Поиск гомологов белков в геноме паразитического грибоподобного нечта
Выполнено локальным бластом на кодомо.
Белочек 1 — сукцинат-дегидрогеназа (цитохром b560) из человеков (C560_HUMAN, Q99643)
Сукцинат-дегидрогеназа является одним из белков дызательной цепи (а конкретно, комплексом 2), очень важна для нормальной жизнедеятельности клетки, так как осуществляет процесс переноса электронов в дыхательной цепи. Комплекс переносит электроны на убихинон и окисляет сукцинат с восстановлением FAD. Если верить логике, должна быть у всех.
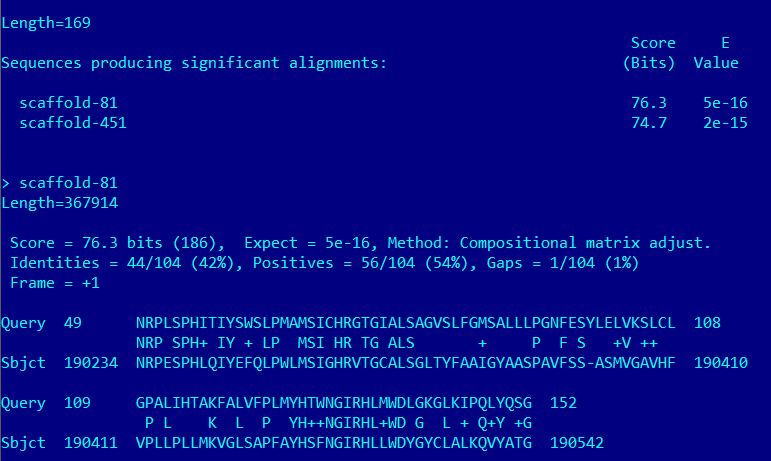
На картинке выше отображен результат работы локального blast. Как видно, находок всего 2, обе с низким E-value, но вместе с тем и низким score. Так как я предполагаю, что сукцинат-дегидрогеназный комплекс распространен среди всех эукариот, а выравнивания не позволяют установить однозначную гомологию, я посчитаю эти находки условно-гомологичными.
Белочек 2 — альфа-актинин из человеков (P35609, ACTN2_HUMAN)
Является одним из белков цитоскелета, а цитоскелет присутствует у всех эукариот, умеет связывать АТР и магний, тем самым умеет катализировать реакцию гидролиза АТР.
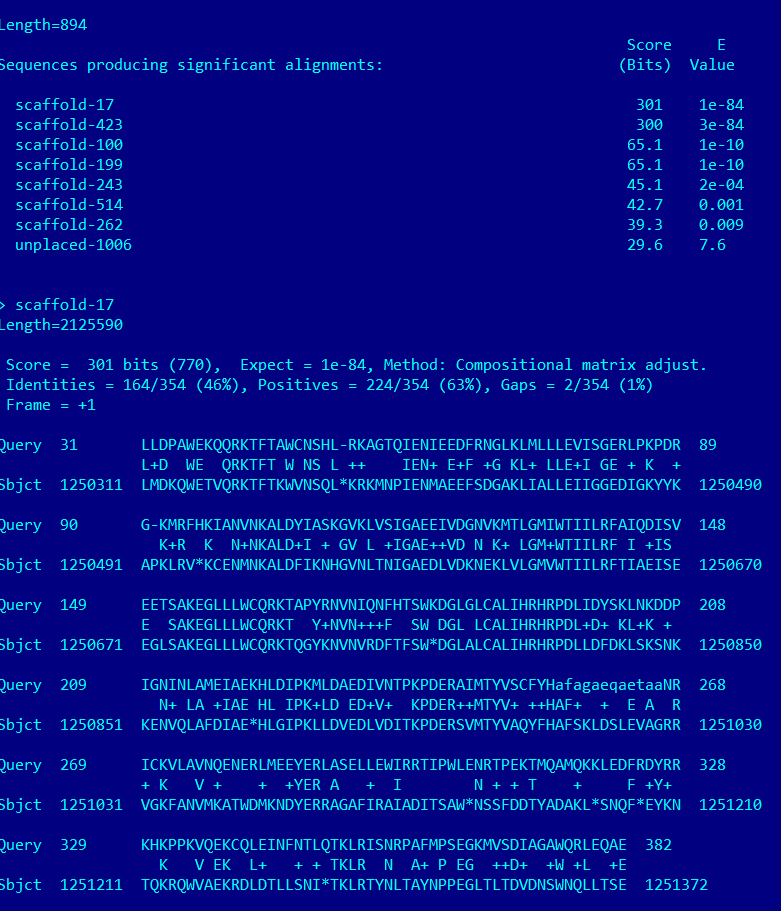
В ходе работы алгоритма были найдены 2 скэффолда, для которых наблюдается достаточно высокий score и низкий E-value. Просмотр выравнивания для первой находки показывет достаточно высокую степень схожести последовательностей (особенно на некоторых участках), что позволяет с высокой вероятностью установить их гомологию. Ура!
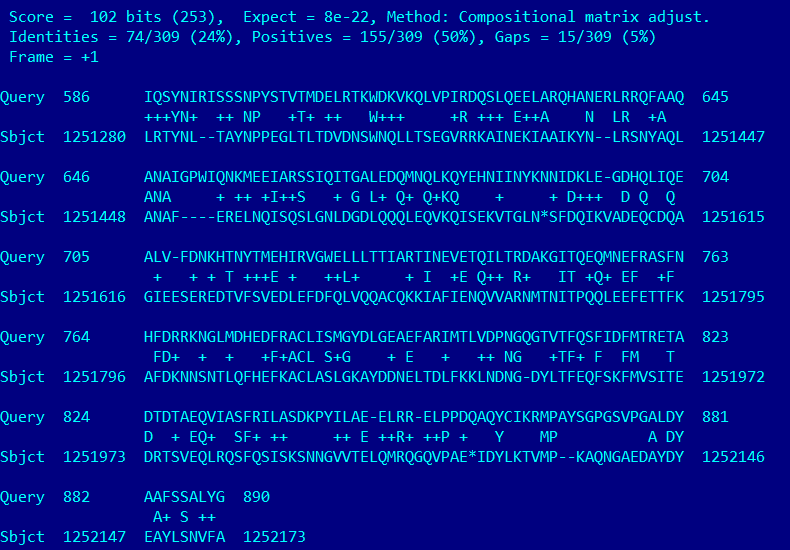
Просмотр второго выравнивания показывает, что вторая часть белка тоже выравнивается с достаточно неплохим покрытием и уровнем схожести. Теперь точно ура актину!
Белочек 3 — тирозин-киназа 2-бета из мышек (Q9QVP9, FAK2_MOUSE)
Эукариотические клетки славятся своими безумными регуляторными каскадами, и сама тирозин-киназа есть их важная часть. Она фосфолирирует ОН-группу тирозина, тем самым изменяя каталитические свойства. Сериновую киназу я посчитала неинтересной, поэтому будет тирозиновая.
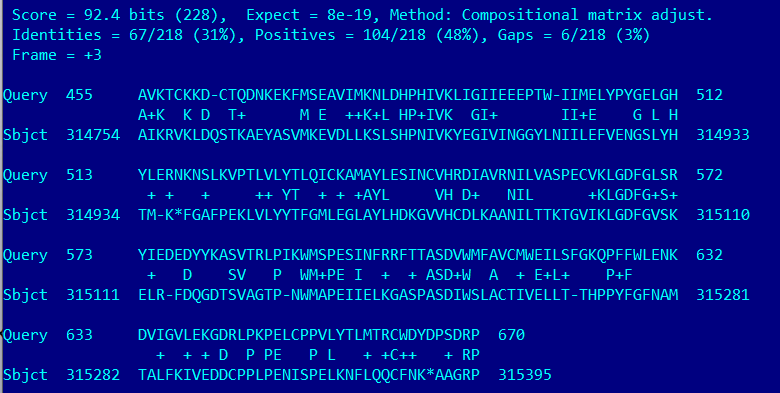
Алгоритм находит множество скэффолдов, у которых все не очень хорошо со score. На картинке представлено лучшее выравнивание, по нему нельзя установить гомологию данных последовательностей. Но вообще, скорее всего у этого грибоподобного паразита есть какая-либо тирозинкиназа, но возможно не из этого подсемейства (она должна быть!).
Поиск гена в контиге
Поиск выполнялся с использованием контига для описанной в предыдущем праке сборке генома. Я взяла самый маленький из имеющихся. Поиск был ограничен по организму: я искала по последовательностям только из Arabidopsis thaliana.
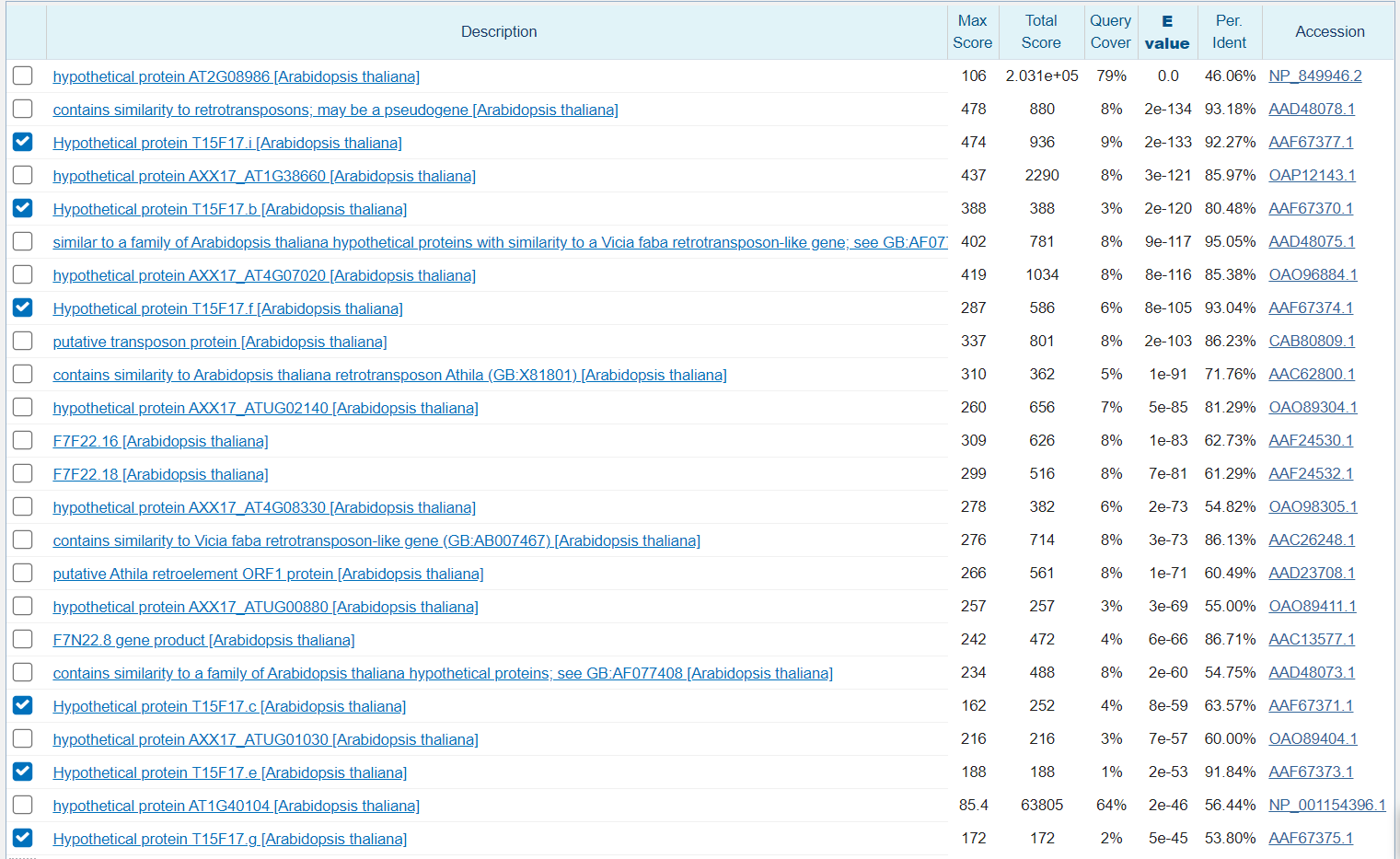
На картинке выше отражена часть выдачи алгоритма blastx. Низкие E-value и высокий процент идентичности позволяют установить наличие гипотетического белка T15F17 в данном контиге в какой-либо его изоформе.
*Сайт был полностью написан в ночь на 29 сентября очень уставшей Лерой. Пожалуйста, простите мне опечатки, как только я все разгребу, я их поправлю!