Алгоритм UPGMA дал укорененные деревья, но неправильно, Nieghbor Joining (NJ)
- неукорененное (впоследствии оказался случайно укорененным).
С помощью программы retree пакета PHYLIP я переукоренила неправильные деревья (метод переукоренеия поддеревьев!).
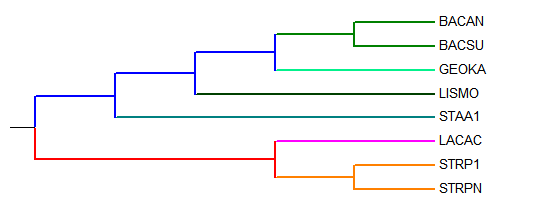
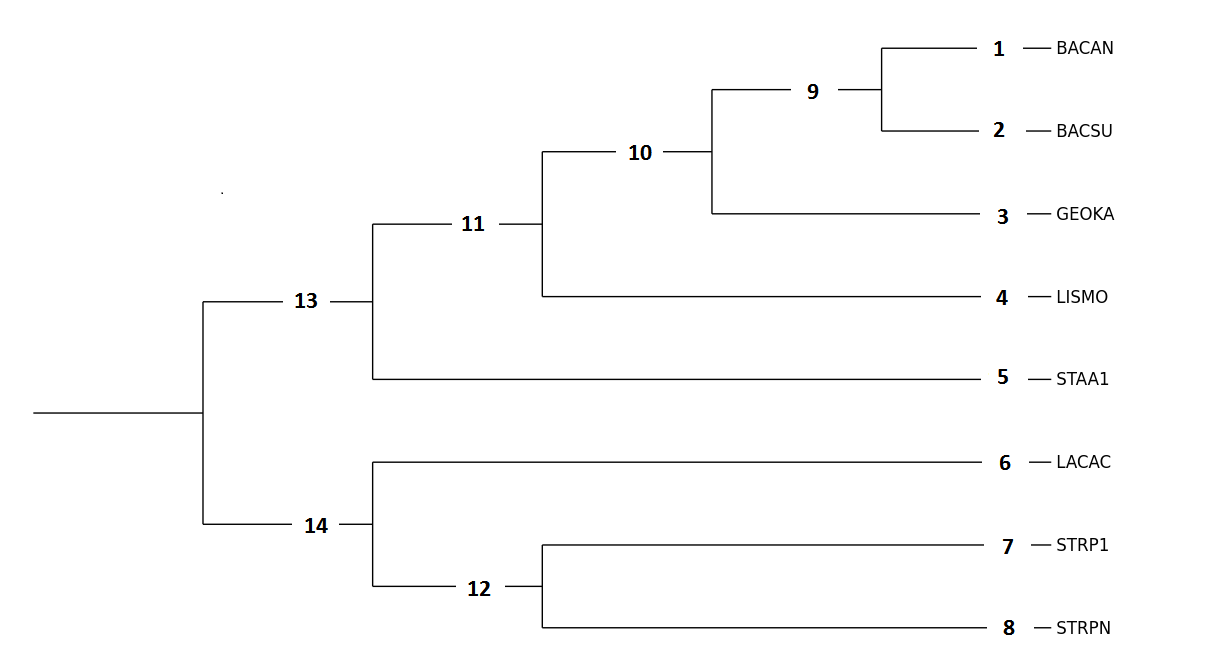
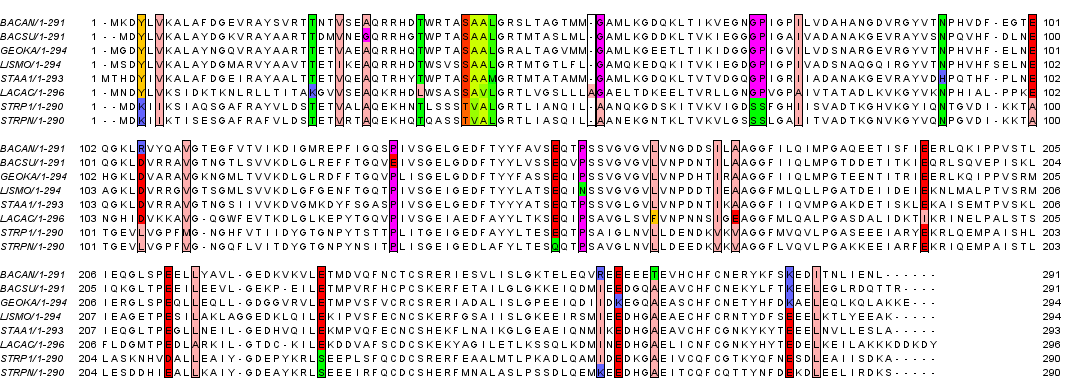
Результаты работы программ, картинкии в последовательности вид JalView; вид MEGA; вид MEGA переукоренное:
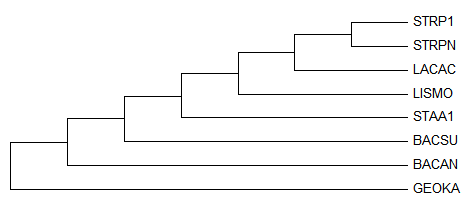
UPGMA Id
Ультраметрическое дерево, предумсотрены молекулярные часы:
Newick format:
((STRPN,STRP1),((BACAN,(((GEOKA,BACSU),STAA1),LISMO)),LACAC));
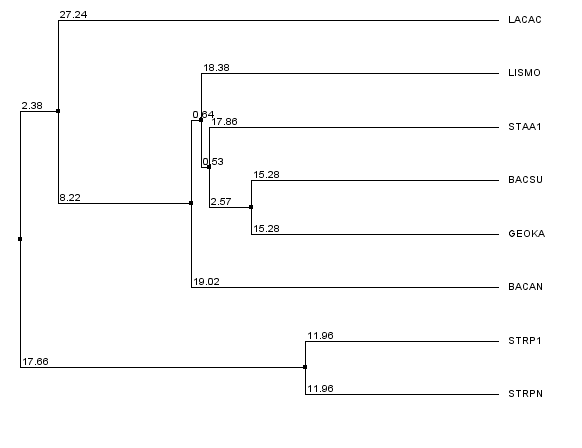
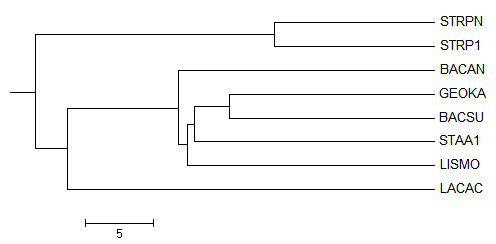
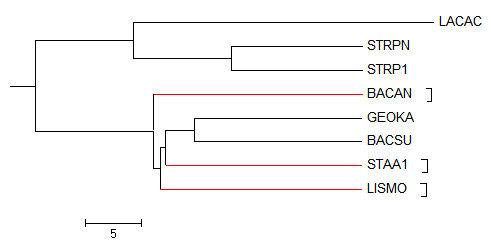
Листья, расположение которых отличается от исходного: BACAN, STAA1, LISMO
В терминах нетривиальных ветвей:
ветви
(GEOKA,BACSU)vs(LACAC,STRPN,STRP1,BACAN,STAA1,LISMO) и
(LACAC,STRPN,STRP1)vs(BACAN,GEOKA,BACSU,STAA1,LISMO)
выделены правильно.
Ветви
(GEOKA,BACSU,STAA1)vs(LACAC,STRPN,STRP1,BACAN,LISMO) и
(GEOKA,BACSU,STAA1,LISMO)vs(LACAC,STRPN,STRP1,BACAN)
выделены неравильно.
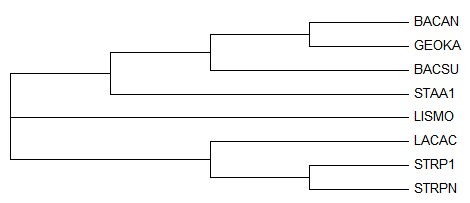
UPGMA BLOSUM62
Newick format:
((STRPN,STRP1),((BACAN,(((GEOKA,BACSU),STAA1),LISMO)),LACAC));
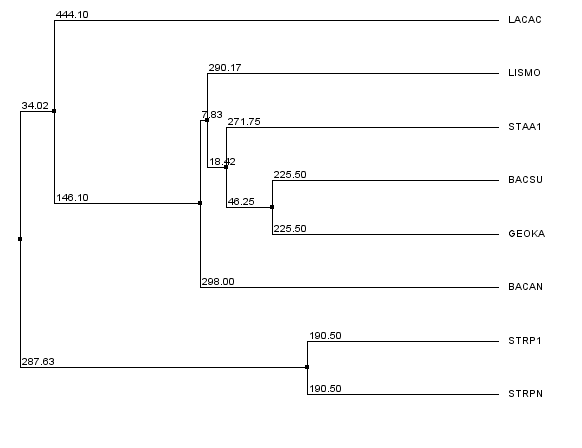
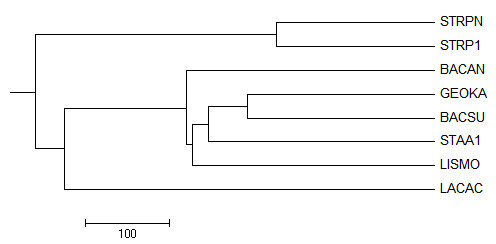
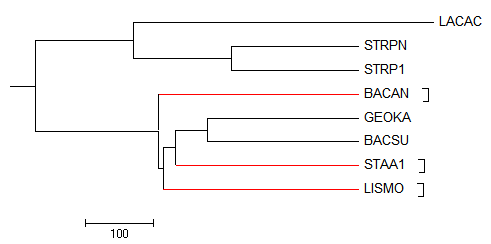
Листья, расположение которых отличается от исходного: BACAN, STAA1, LISMO
В терминах нетривиальных ветвей:
ветви
(GEOKA,BACSU)vs(LACAC,STRPN,STRP1,BACAN,STAA1,LISMO) и
(LACAC,STRPN,STRP1)vs(BACAN,GEOKA,BACSU,STAA1,LISMO)
выделены правильно.
Ветви
(GEOKA,BACSU,STAA1)vs(LACAC,STRPN,STRP1,BACAN,LISMO) и
(GEOKA,BACSU,STAA1,LISMO)vs(LACAC,STRPN,STRP1,BACAN)
выделены неравильно.
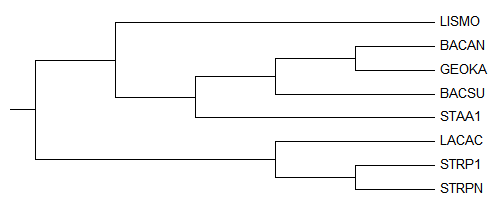
Neighbor Joining Id
Неультраметрическое дерево, не предусмотрены молекулярные часы:
Newick format:
((GEOKA,(BACSU,(STAA1,(LISMO,(LACAC,(STRPN,STRP1)))))),BACAN);
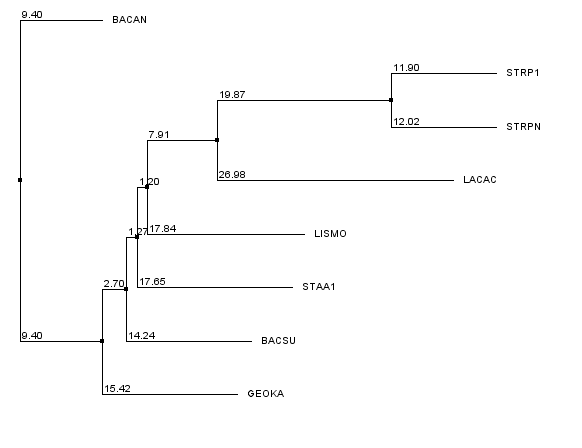
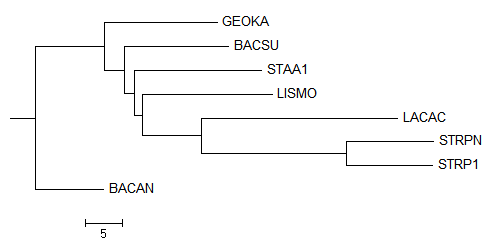
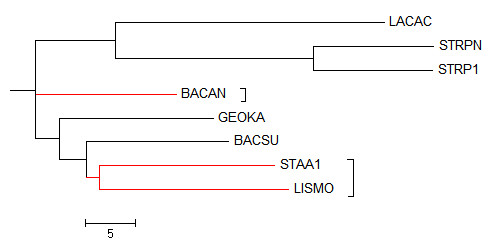
Листья, расположение которых отличается от исходного: BACAN, STAA1, LISMO
В терминах нетривиальных ветвей:
ветвь
(LACAC,STRPN,STRP1)vs(BACAN,GEOKA,BACSU,STAA1,LISMO)
выделена правильно.
Ветви
(LACAC,STRPN,STRP1,BACAN)vs(BACAN,GEOKA,BACSU,STAA1,LISMO)
(LISMO,BACSU,STAA1)vs(LACAC,STRPN,STRP1,BACAN,GEOKA) и
(STAA1,LISMO)vs(GEOKA,BACSU,LACAC,STRPN,STRP1,BACAN)
выделены неравильно.
Neighbor BLOSUM62
Newick format:
((GEOKA,(BACSU,(STAA1,(LISMO,(LACAC,(STRPN,STRP1)))))),BACAN);
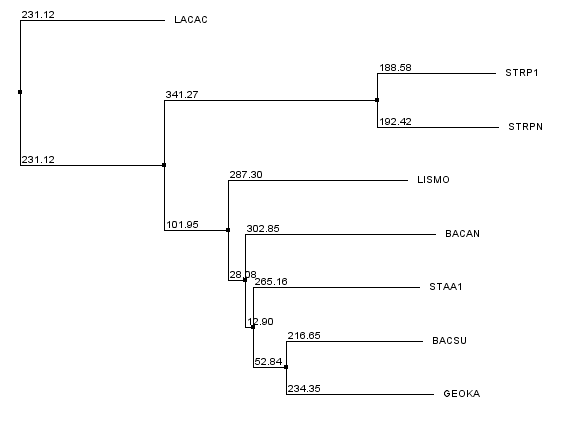
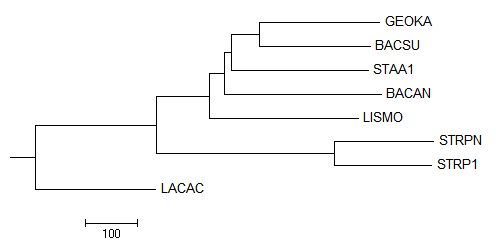
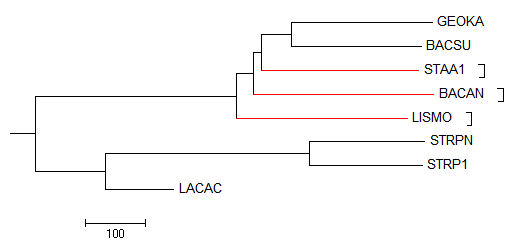
Листья, расположение которых отличается от исходного: BACAN, STAA1, LISMO
В терминах нетривиальных ветвей:
ветви
(GEOKA,BACSU)vs(LACAC,STRPN,STRP1,BACAN,STAA1,LISMO) и
(LACAC,STRPN,STRP1)vs(BACAN,GEOKA,BACSU,STAA1,LISMO)
выделены правильно.
Ветви
(GEOKA,BACSU,STAA1)vs(LACAC,STRPN,STRP1,BACAN,LISMO) и
(GEOKA,BACSU,STAA1,BACAN)vs(LACAC,STRPN,STRP1,LISMO)
выделены неравильно.
Пояснение. Для описания различий построенного дерева от реальной таксономи я специально выбрала листья,
рассматривая их как поддеревья, присоединенные к данной нетривиальной ветви.
Например, BACAN во всех случаях был присоединен не так глубоко в поддереве Bacillales, как в дереве таксономии.
В случае UPGMA это означает,
что по белку-шаперонину Bacillus anthracis почему-то ближе к Lactobacillales, чем остальные представители
Bacillales (точнее, далек от Lactobacillales, как и от Bacillales).
Для случая NJ нет ветви, объединяющей BACAN и Lactobacillales, но и нет ветви, объединяющей обоих Bacillus.
Neighbor Joining % Identity расположил LISMO и STAA1 глубже в поддереве Bacillales, чем они есть на самом деле.
Адрес сервиса: http://www.genebee.msu.ru/services/phtree_reduced.html
Интерфейс сервиса:
На вход запрашивает выравнивание, но не в формате fasta. Пример формата выравнивания - здесь же, на странице.
Результат с параметрами по умолчанию (без бутстрепа, выходное дерево в формате PHYLIP,
матрица BLOSUM62, учет только гомологичных участков выравнивания, алгоритмы и кластерный, и топологический):
PHYLOGENETIC TREE
CLUSTER ALGORITHM
0.538685
_____________________________________________________________________ STRP1
| |___________________________ STRPN
|____________________________________________________________________ LACAC
|_______________________________________________________________ LISMO
| |__________________________________________ STAA1
| |_______________________________________ BACSU
| |_________________________________ GEOKA
|____________________________________________ BACAN
* The phylogenetic tree in Phylip format
((STRP1:0.213000,STRPN:0.213000):0.325685,(LACAC:0.495293,((LISMO:0.325846,(STAA1:0.304137,(BACSU:0.256374,GEOKA:0.256374):0.047763):0.021708):0.018953,BACAN:0.344799):0.150494):0.043391);
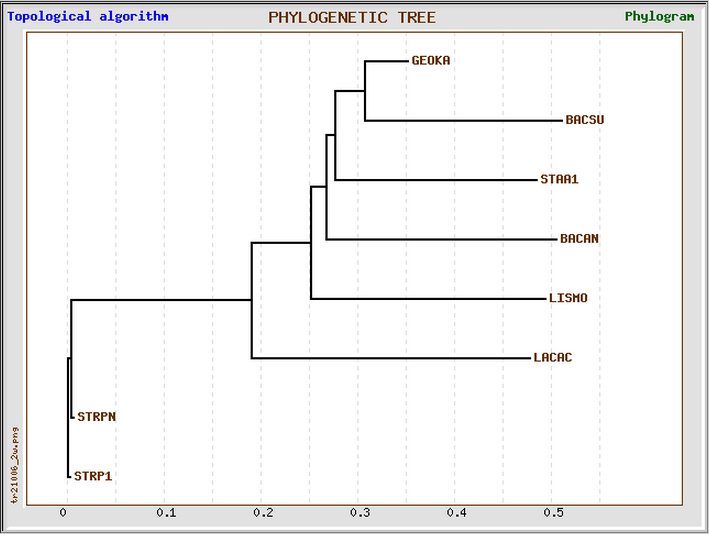
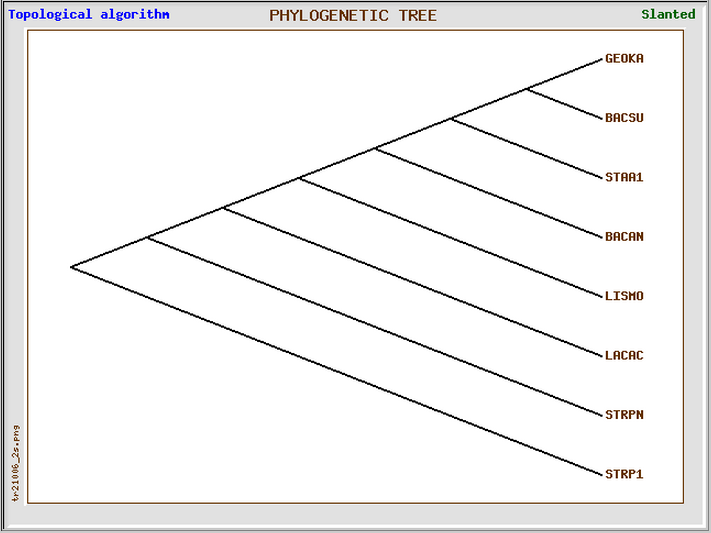
TOPOLOGICAL ALGORITHM
_______________________________________________ GEOKA
| | | || |___________________________ BACSU
| | | ||____________________________ STAA1
| | | |________________________________ BACAN
| | |________________________________ LISMO
| |_______________________________________ LACAC
| STRPN
| STRP1
* The phylogenetic tree in Phylip format
(((((((GEOKA:0.046229,BACSU:0.204808):0.031303,STAA1:0.210120):0.009451,BACAN:0.239592):0.016229,LISMO:0.244173):0.061712,LACAC:0.289526):0.186829,STRPN:0.001000):0.000500,STRP1:0.000500);
Distance Matrix
1 2 3 4 5 6 7 8
1 STRP1 0.000 0.213 0.554 0.518 0.524 0.516 0.531 0.551
2 STRPN 0.213 0.000 0.554 0.527 0.518 0.509 0.551 0.548
3 LACAC 0.554 0.554 0.000 0.470 0.506 0.493 0.506 0.510
4 LISMO 0.518 0.527 0.470 0.000 0.360 0.321 0.329 0.333
5 BACAN 0.524 0.518 0.506 0.360 0.000 0.338 0.330 0.311
6 STAA1 0.516 0.509 0.493 0.321 0.338 0.000 0.275 0.333
7 BACSU 0.531 0.551 0.506 0.329 0.330 0.275 0.000 0.256
8 GEOKA 0.551 0.548 0.510 0.333 0.311 0.333 0.256 0.000
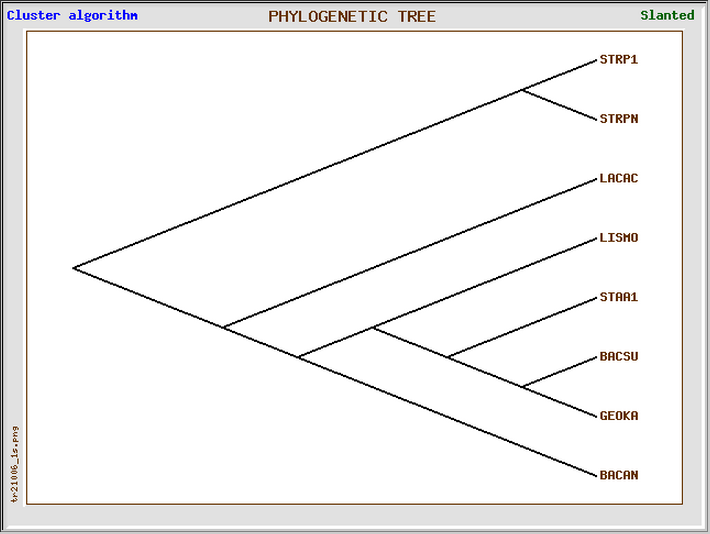
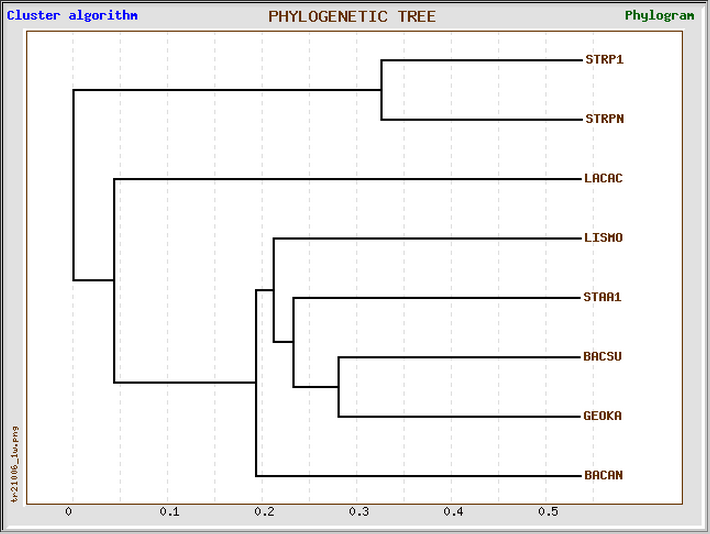
Изображения деревьев в прмоугольной (слева) и угловой (справа) формах:
Кластерным методом:
Топологическим алгоритмом: