Цель данного практикума - подготовить необходимые файлы, изучить качество чтений, отфильтровать чтения и
подготовить их для картирования на одну из хромосом (мне досталась 8 хромосома).
1. Описание файла с референсом.
Файл chr8.fna с референсом восьмой хромосомы оказался fasta-файлом с её последовательностью. В первой
строке дана общая информация о последовательности: RefSeq AC (NC_000008.11) и описание с указанием организма
и названием геномной сборки (Homo sapiens chromosome 8, GRCh38.p13 Primary Assembly). В начале и в конце
последовательности идут достаточно протяженные участки неизвестных нуклеотидов (N).
Первая строка выглядит так:
>NC_000008.11 Homo sapiens chromosome 8, GRCh38.p13 Primary Assembly
2. Индексация референса.
BWA - это программный пакет для картирования малоразличающихся последовательностей относительно большой
референсной последовательности (в моем случае 8 хромосома человека). Он состоит из трех алгоритмов:
BWA-backtrack,
BWA-SW и
BWA-MEM [1]. В данном практикуме мы используем алгоритм BWA-SW. Сначала нужно
проиндексировать референс с помощью команды bwa index -a bwtsw
chr8.fna. Команда index производит саму индексацию, опция -a позволяет указать
название алгоритма, для которого производится индескация (bwtsw), chr8.fna - имя
индексируемого файла.
На выходе программа выдает пять файлов: chr8.fna.amb, chr8.fna.ann, chr8.fna.bwt, chr8.fna.pac и chr8.fna.sa.
Файлы chr8.fna.bwt, chr8.fna.pac и chr8.fna.sa в текстовом редакторе просмотреть не удалось. В файле
chr8.fna.amb видимо содержится информация о неопознанных нуклеотидах, а в файле chr8.fna.ann общая
информация о данной последовательности.
3. Описание образца.
В базе NCBI по данному ID
SRR10720419 была найдена некоторая
информацио об образце (представлена в таблице 1).
Для проверки качества двух парноконцевых чтений была запущена команда fastqc для каждого файла:
fastqc SRR10720419_1.fastq.gz
fastqc SRR10720419_2.fastq.gz
На выходе программа выдает файлы SRR10720419_1_fastqc.html и SRR10720419_2_fastqc.html. Из них была получена
следующая информация:
Количество пар чтений в каждом из файлов получилось 41277367. Это совпадает с ожидаемым числом,
указанным в NCBI (см пункт 3).
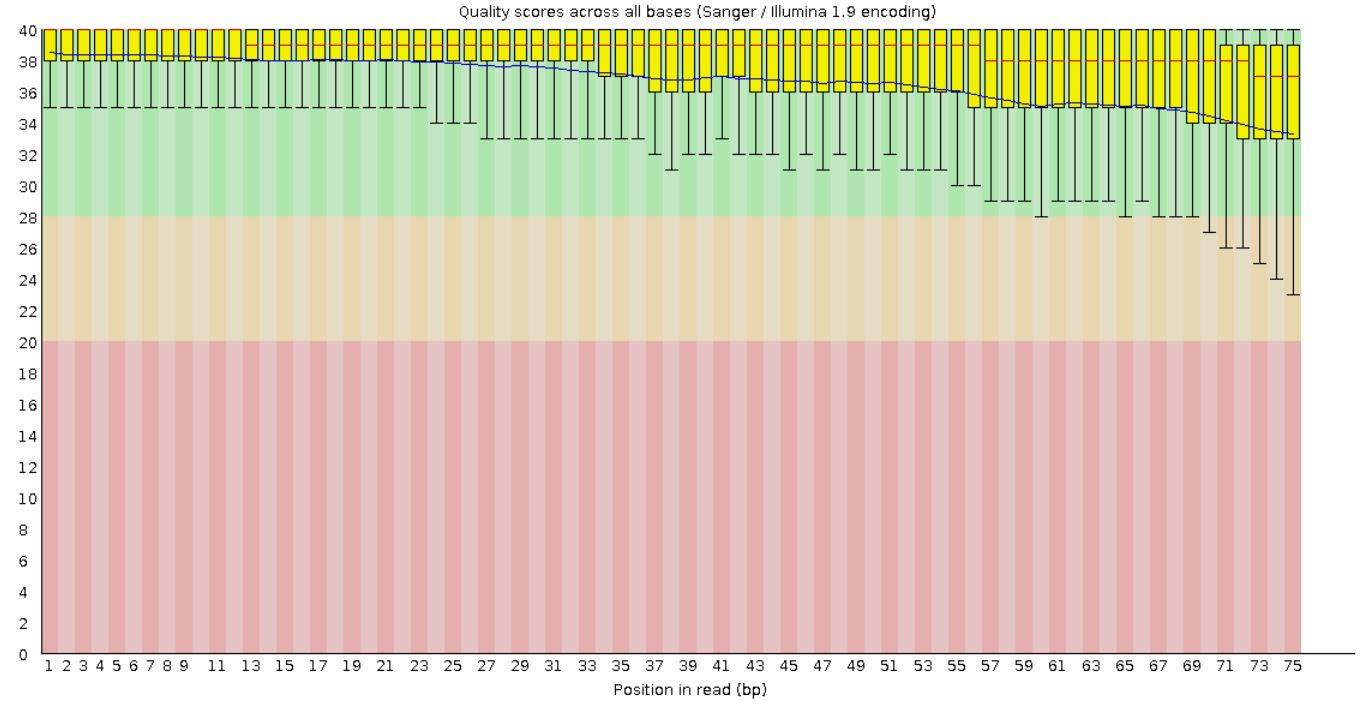
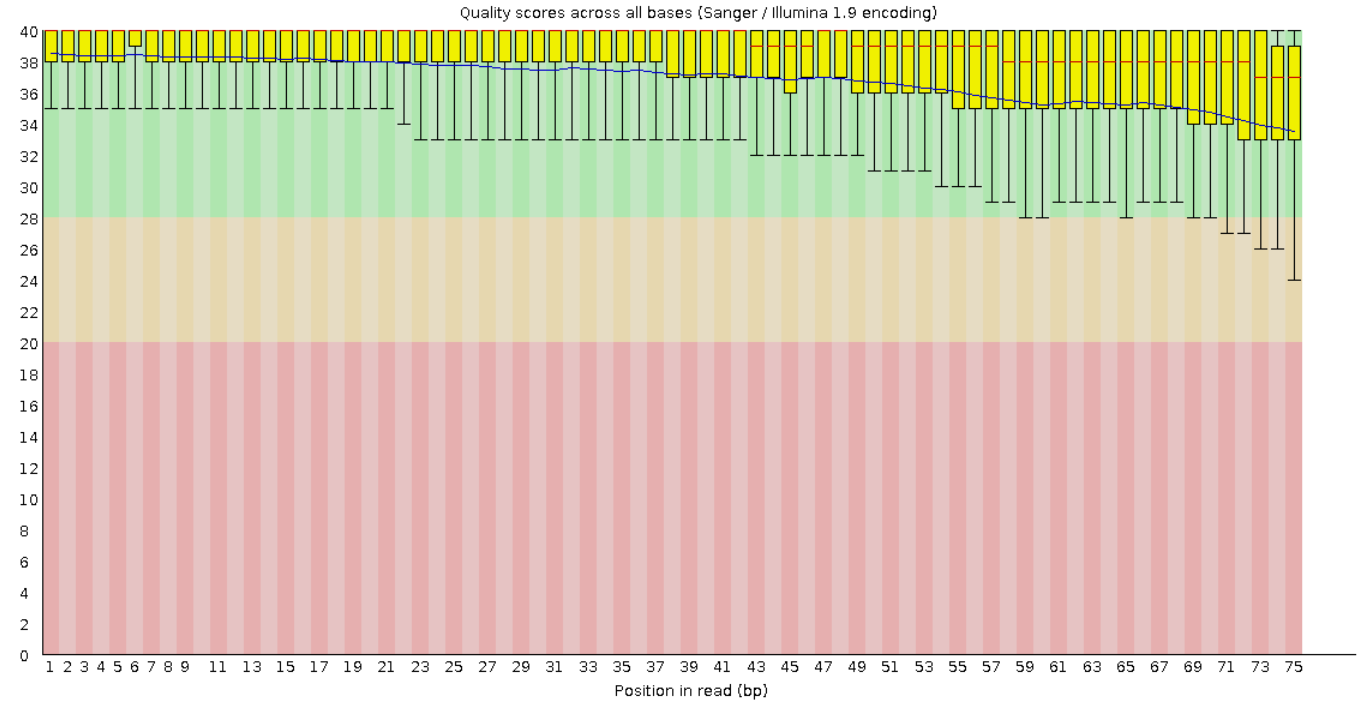
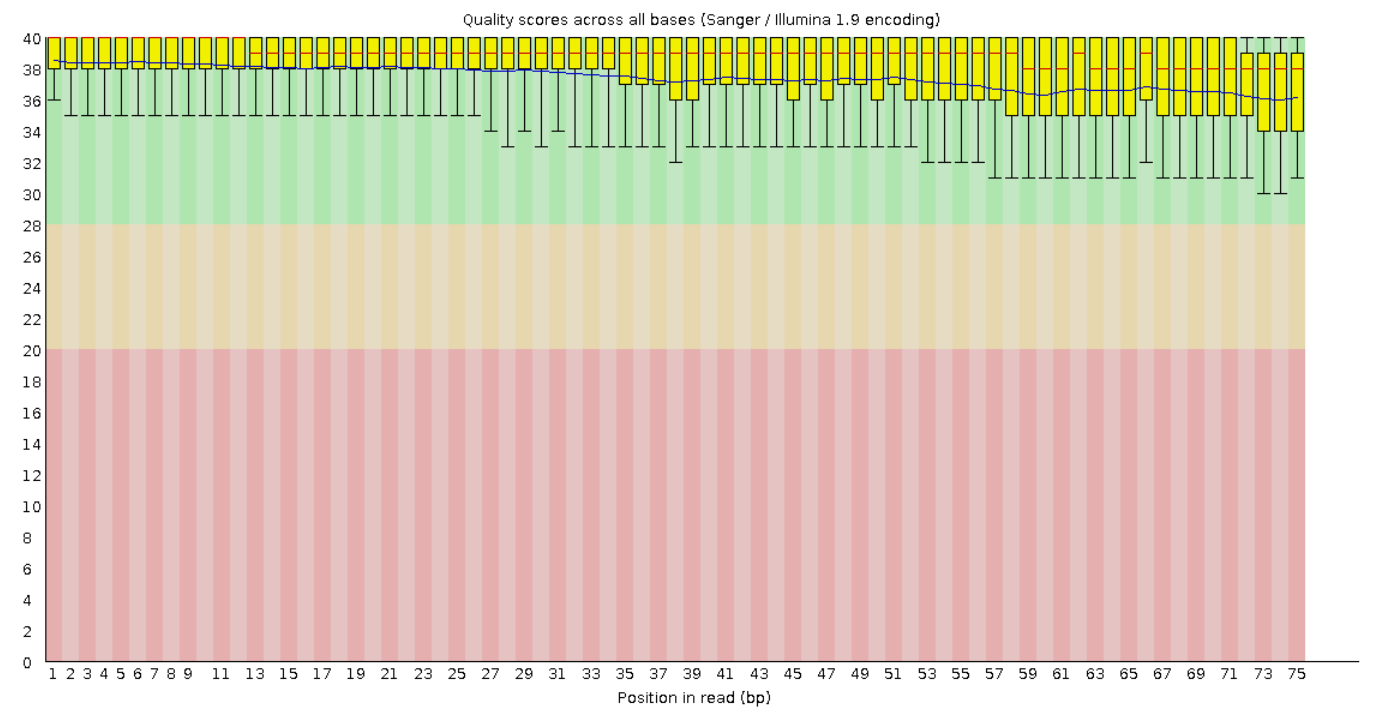
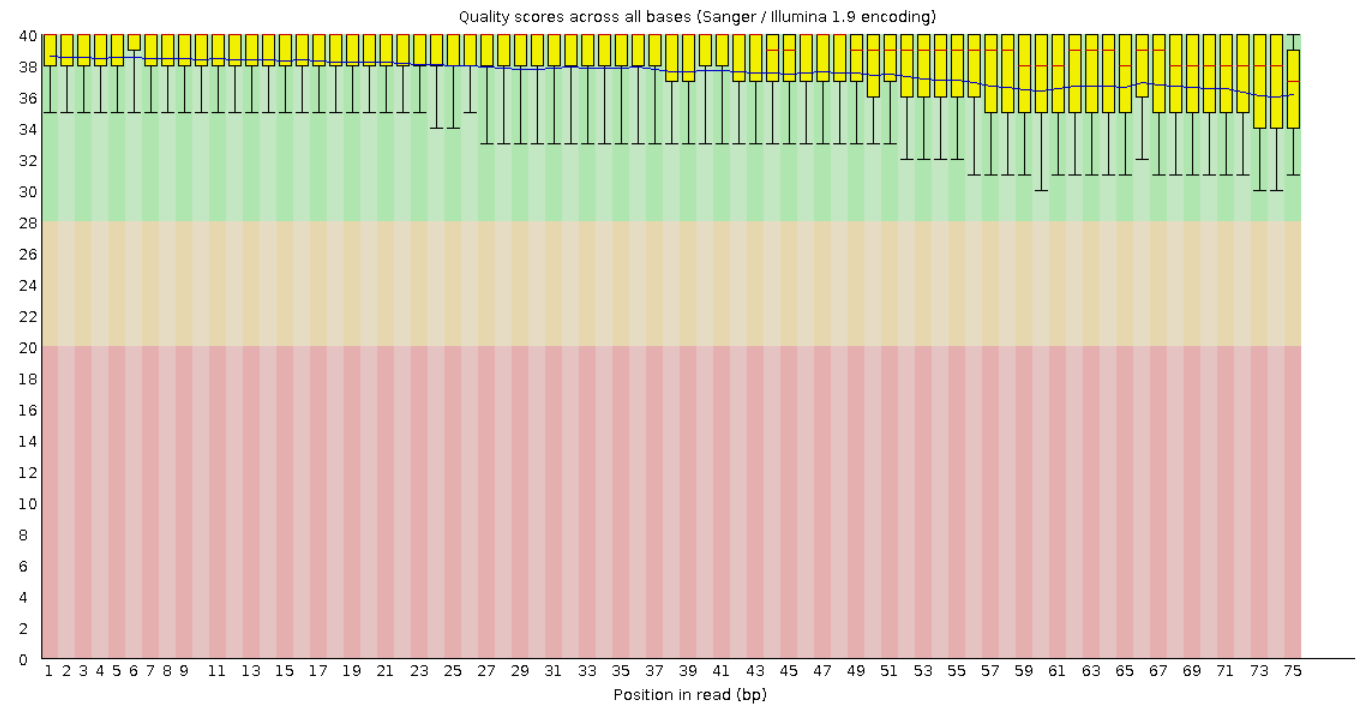
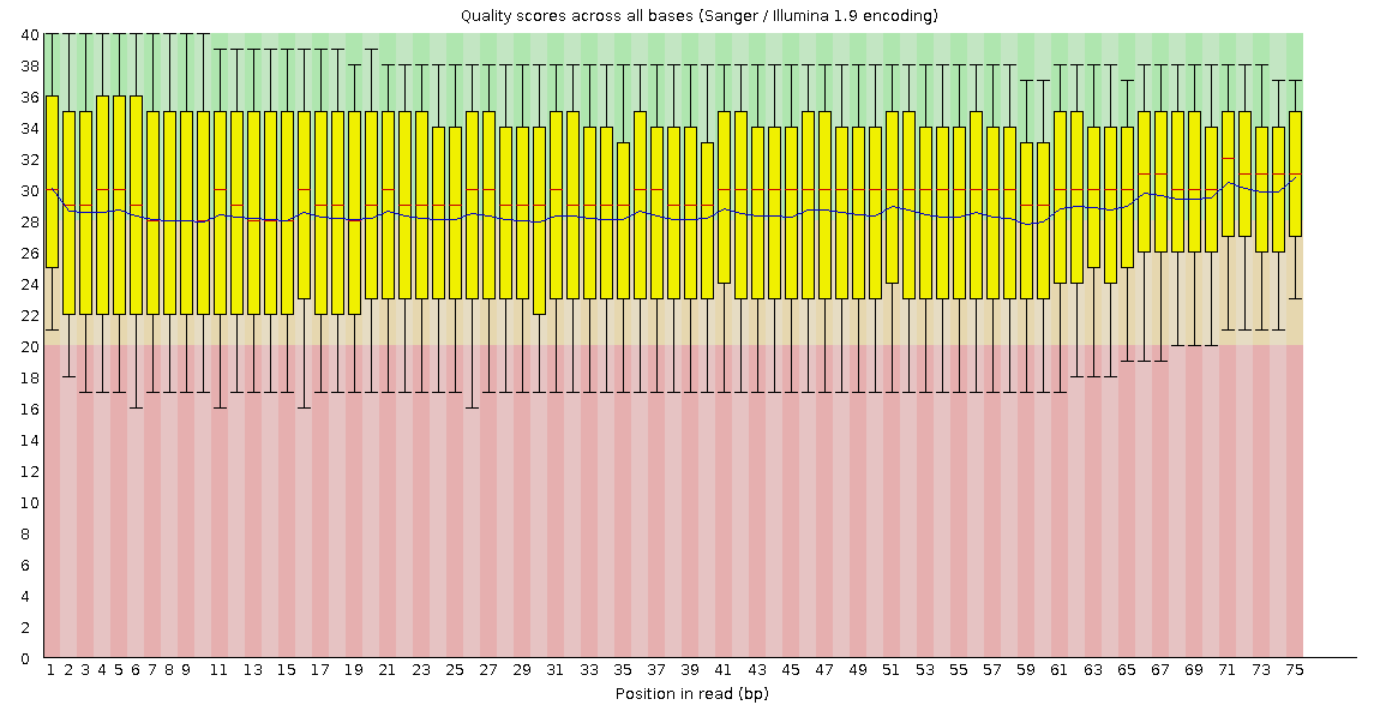
На рисунках 1 и 2 представлены графики Per base sequence quality для прямого и обратного чтения
соответственно. Из графиков видно, что для каждой позиции (они отложены по оси x) Phred Score больше 20.
А значения большие 20 обеспечивают 99% вероятность правильного распознавания нуклеотида. Также видно,
что качество чтения к концу фрагментов в среднем ухудшается.
Рис. 1. Per base sequence quality для "прямого
чтения".
Рис. 2. Per base sequence quality для "обратного
чтения".
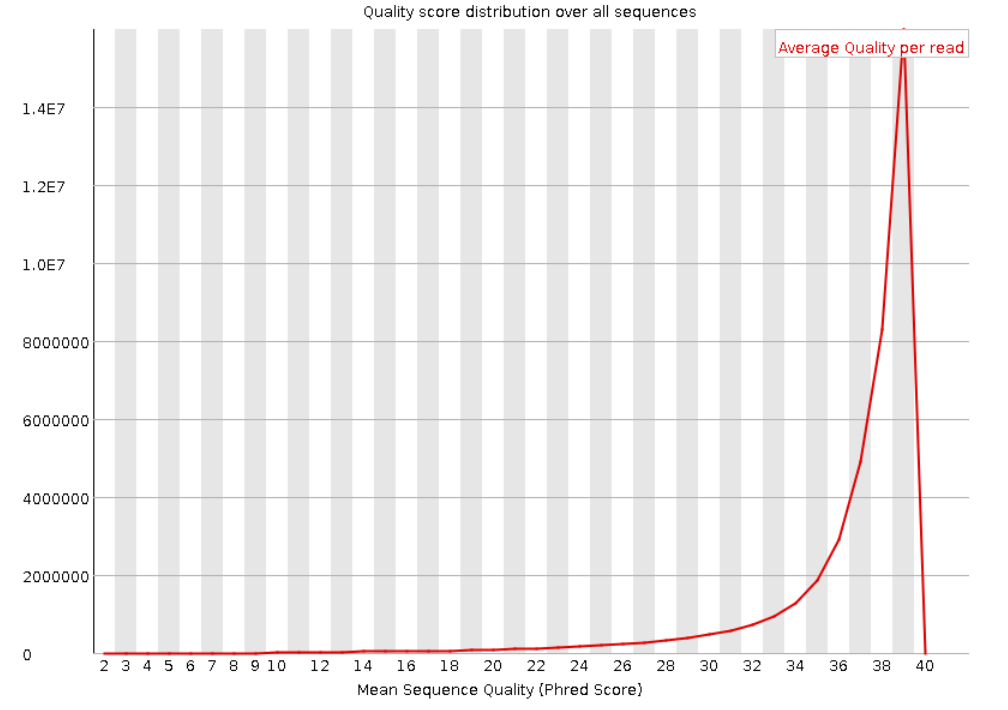
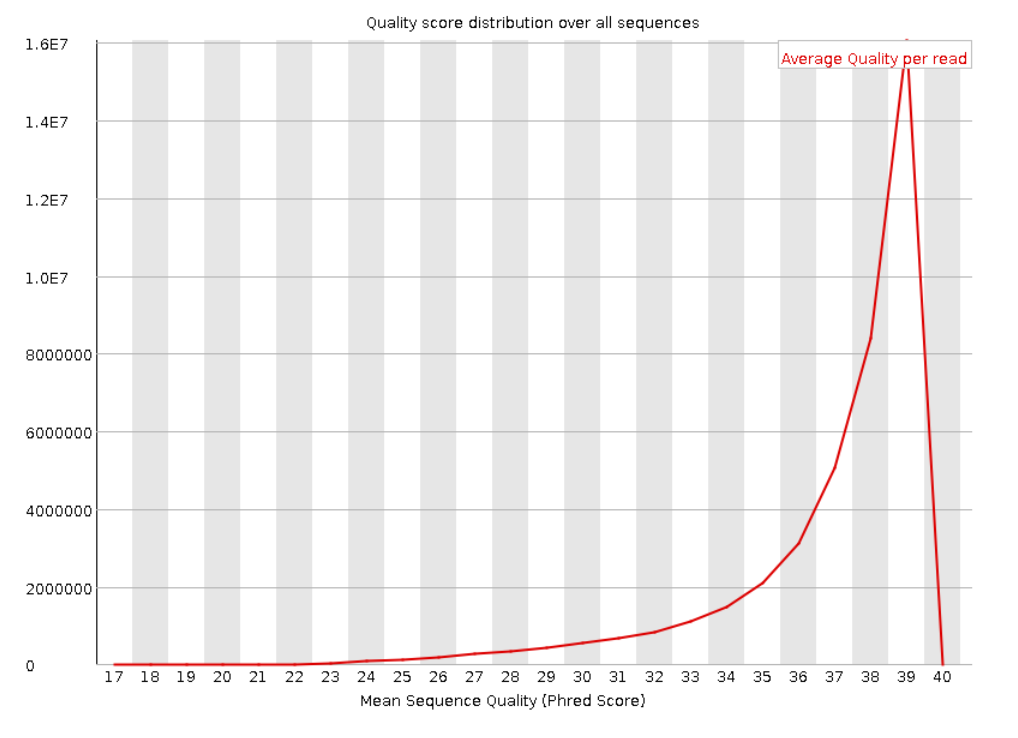
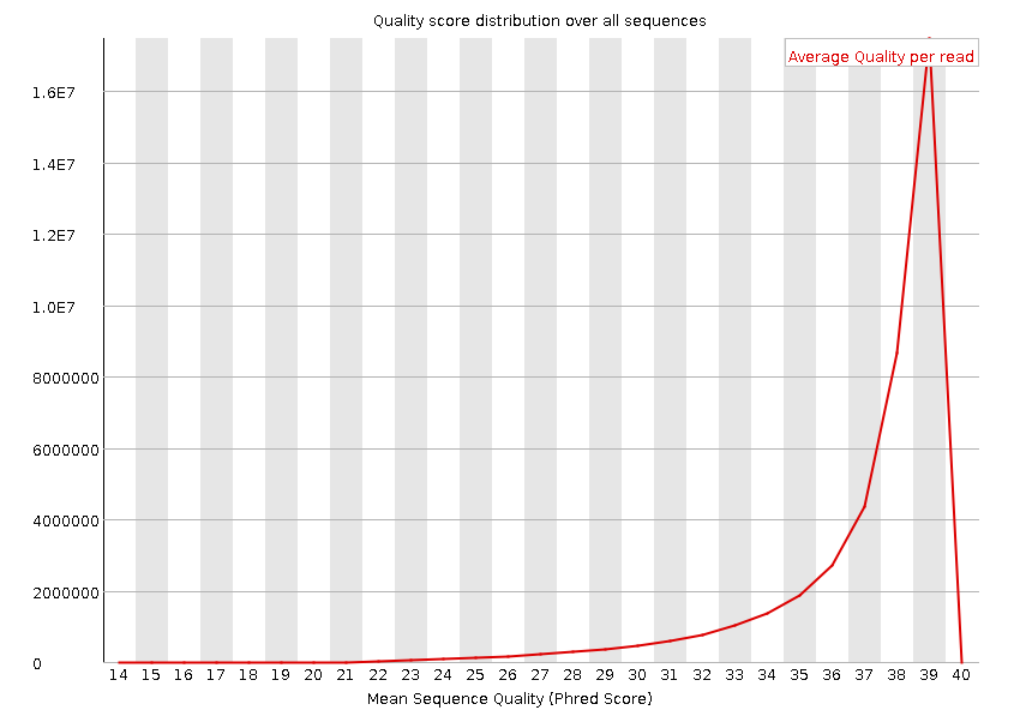
Ниже представлены графики распределения Phred Score по всем прочтениям. Видно, что для прямых и обратных
прочтений в среднем значение равно 39. А чтений с Phred Score меньше 20 очень мало. Это говорит о
высоком качестве наших данных.
Рис. 3. Per sequence quality scores для "прямого
чтения".
Рис. 4. Per sequence quality scores для "обратного
чтения".
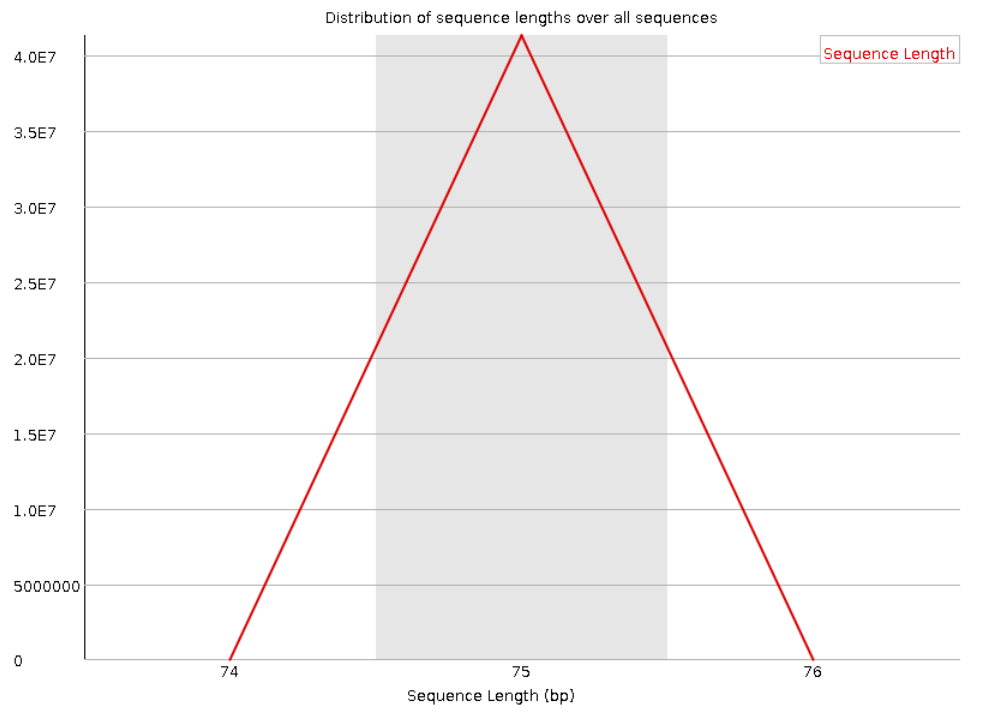
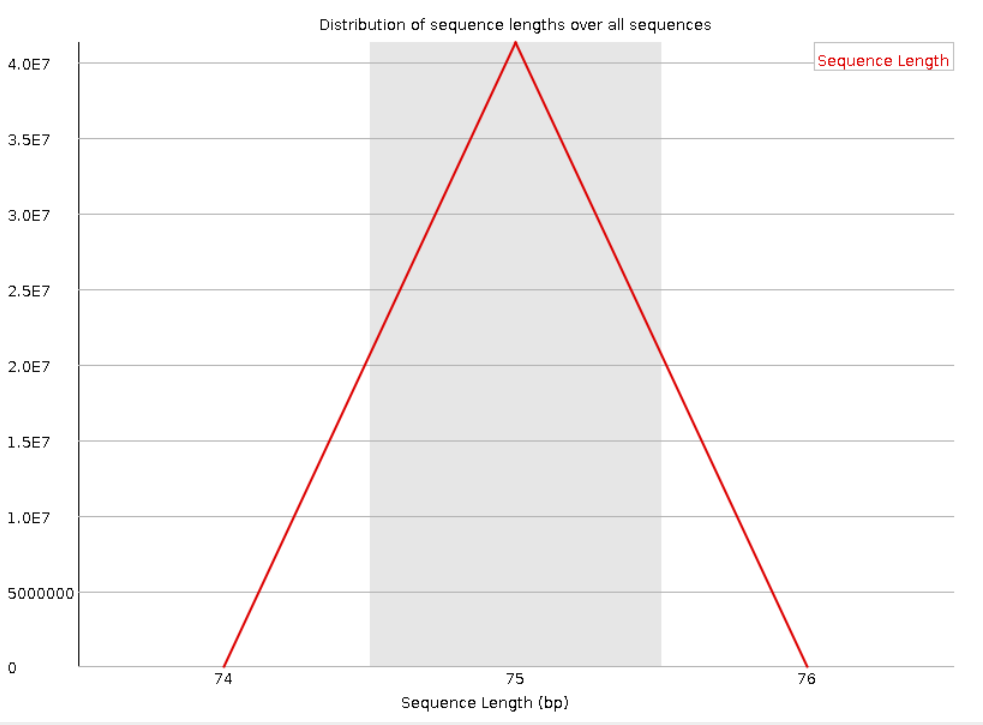
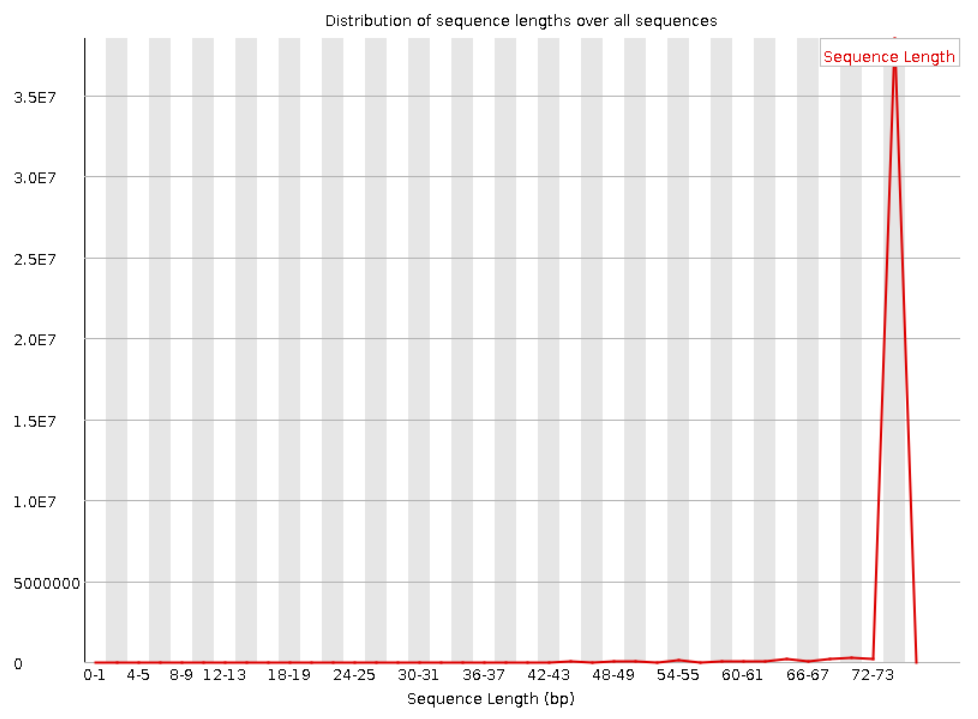
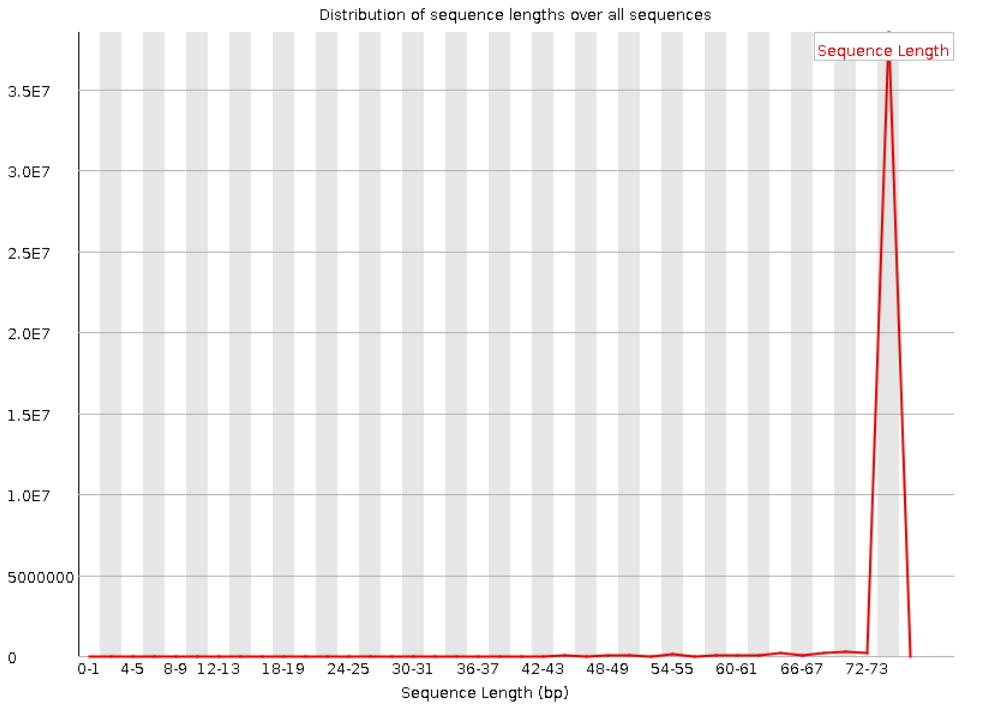
Следующая пара графиков показывает распределение длин прочтений. Видно, что для прямого и обратного
чтения графики совпадают. Также видно, что все прочтения имеют длину в 75 нуклеотидов.
Рис. 5. Sequence Length Distribution для "прямого
чтения".
Рис. 6. Sequence Length Distribution для "обратного
чтения".
5. Фильтрация чтений.
Далее с помощью программы Trimmomatic наши чтения были дополнительно отфильтрованы. В командную строку была
введена следующая команда:
java -jar /usr/share/java/trimmomatic.jar - запуск программы на kodomo
PE - Paired End Mode. Режим trimmomatic, при котором фильтруется пара прямого и обратного чтения
(на вход подается два файла). На выходе получается 4 файла: два "парных", которые получены с помощью
обработки обоих чтений; и два "непарных", где каждый фильтровался по отдельности соответственно.
-threads 12 - были задействованы 12 ядер для вычислений, а не все ядра узла.
-phred33 - кодировка Phred Score.
TRAILING:20 - выкидывает нуклеотиды, чей Phred Score меньше 20.
MINLEN:50 - удаляет последовательности длины меньше 50.
6. Проверка качества триммированных чтений.
Далее к четырем получившимся файлам была снова применена команда fastqc. Ниже представнены вводимые команды и
анализ получившихся файлов:
Количество пар чтений (для paired) стало 41170670. Это составляет 99.74% от количества чтений до
фильтрации.
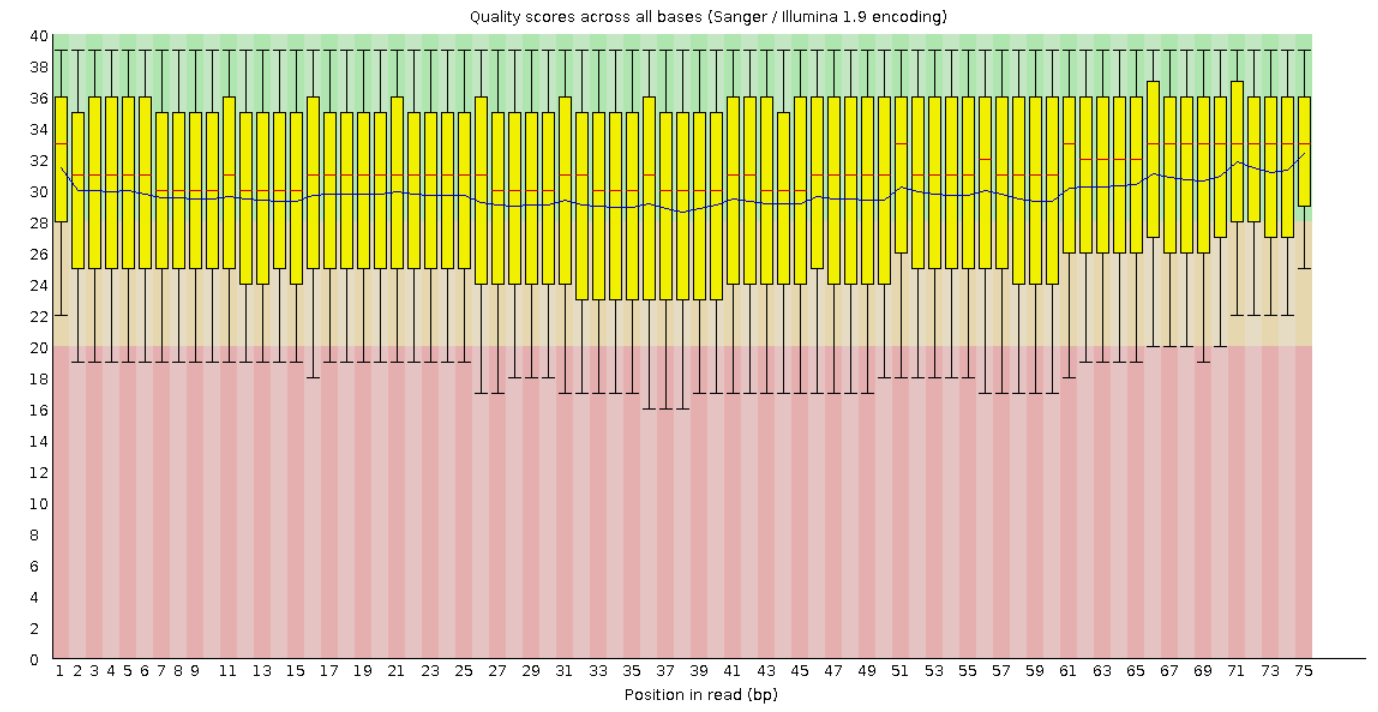
Ниже представлено четыре графика для соответствущих файлов с оценкой Phred Score по позициям. Видно, что
для paired качество прочтений улучшилось относительно качества чтений до фильтрации. Для unpaired
наоборот значительно ухудшилось - по многим позициям Phred Score уходит в красную зону, а значит
вероятность ошибки в таких позициях достаточно велика.
Рис. 7. Per base sequence quality для парного
"прямого"
чтения.
Рис. 8. Per base sequence quality для парного
"обратного"
чтения.
Рис. 9. Per base sequence quality для непарного
"прямого"
чтения.
Рис. 10. Per base sequence quality для непарного
"обратного"
чтения.
На рисунках 11 и 12 представлены распределения Phred Score относительно числа прочтений (для paired).
Видно, что теперь графики начинаются со значений 17 и 14 (до фильтрации с 2 оба). Это говорит о том, что
прочтения с высокой вероятность ошибок были удалены. Но среднее значение (39) осталось прежним.
Рис. 11. Per sequence quality scores для парного
"прямого"
чтения.
Рис. 12. Per sequence quality scores для парного
"обратного"
чтения.
На графиках распределения длин прочтений появились последовательности с длиной сильно меньше 75.
Вероятно во время фильтрации из некоторых последовательностей были удалены нуклеотиды с маленьким Phred
Score. Соответственно уменьшилась длина прочтения и увеличился ее Phred Score. Но большинство прочтений
все еще остались длиной в 75 нуклеотидов (пик на картинках 13 и 14).
Рис. 13. Sequence Length Distribution для парного
"прямого"
чтения.
Рис. 14. Sequence Length Distribution для парного
"обратного"
чтения.