Цель практикума: проанализировать одноконцевые чтения RNA-seq.
1. Подготовка референса.
Создана папка hisat2, в которую была скопированна последовательность 8 хромосомы chr8.fna. Проиндексируем наш
референс с помощью команды: hisat2-build chr8.fna chr8.
Команде hisat2-build на вход был подан файл с референсом (в формате фаста), на выходе получили 8
файлов (chr8.1.ht2, chr8.3.ht2,
chr8.3.ht2, chr8.4.ht2, chr8.5.ht2, chr8.6.ht2, chr8.7.ht2, chr8.8.ht2).
2. Оценка качества чтений.
Далее была создана папка rna-seq. В нее был скопирован файл SRR2015718_1.fastq.gz с одноконцевыми чтениями.
Проанализируем качество исходных чтений с помощью команды cp
SRR2015718_1_fastqc.html ~/public_html/term3/block3/pr14
Всего получилось 33065729 чтений. Картинка из раздела Per base sequence quality и картинка из Length
Distribution представлены ниже:
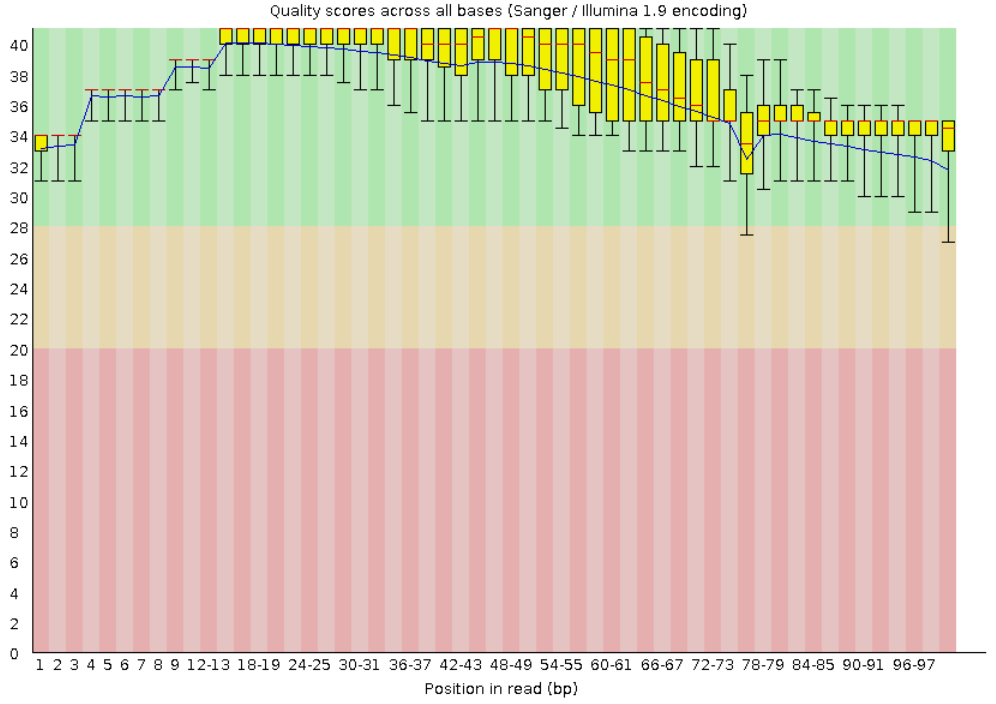
Рис. 1. Per base sequence quality.
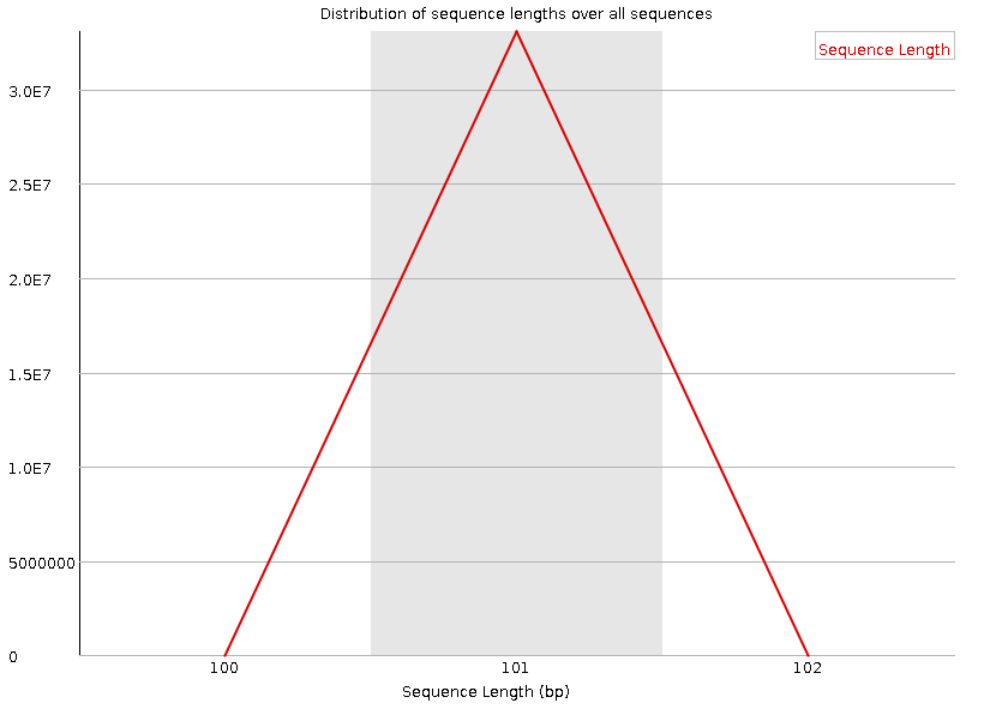
Рис. 2. Length Distribution.
Видно, что каждая позиция нуклеотида находится в зеленой зоне - то есть качество прочтений нас в принципе
устраивает. Из графика распределения длин видно, что каждое чтение имеет длину в 101 нуклеотид.
-p - для данного процесса используем 10 ядер для ускорения.
-x - задание референса.
-k - максимально число выравниваних, имеющих вес больший или равный весу другого
ввыравнивания.
-U - задаем риды.
Получившийся log-файл находится тут. Из него узнаем, что на
нашу хромосому было картировано
3.82% всех ридов.
Далее певедем sam-файл в bam-файл:
samtools sort -o mapped_reads_rna.bam mapped_reads_rna.sam. И отберем чтения только картированные
на нашу хромосому:
samtools index mapped_reads_rna.bam - индексирование
Скопируем к себе в папку файл с разметкой генов для 8-ой хромосомы - gencode.chr8.gtf
Формат gtf состоит из заголовка и тела. Заголовок содержит общую информацию об источнике данных из файла, а
тело состоит 9 столбцов. Из названия и описания представлены в таблице 1 ниже.
Таблица. 1. Информация об образце.
Название столбца
Описание
seqname
Название последовательности, в которой содержится аннотированный ген
source
Источник аннотации
feature
Особенности гена
start
Начало гена
end
Конец гена
score
Вес
strand
Указание цепи
frame
Сдвиг рамки считывания
attribute
Дополнительная информация об аннотации
Далее с помощью команды htseq-count для каждого гена из разметки посчитаем число картированных чтений
на этот ген: htseq-count -f bam -s no -t exon reads_chr8.bam
gencode.chr8.gtf -o htseq-count_chr8.sam 1> htseq_chr8.txt 2> log_htseq.txt.
-f - задание формата (в нашем случае bam).
-t - задает что брать из gtf файла (в нашем случае экзоны).
-s - информация о цепи (в нашем случае отсутствует).
Получим некоторую информацию о получившемся файле: wc -l
htseq_chr8.txt | head -n $lines-5 htseq_chr8.txt | awk '{s+=$2}END{print s}'. Таким образом
в границы генов попало 949398 чтений.