Исследование структуры тРНК
- Краткое описание структуры в файле 1n78.pdb В файле приведены координаты атомов следующих молекул
выделенная из Thermus thermophilus глутамат-тРНК-синетаза.
Для исследования была выбрана цепь С, представляющая глутамат-тРНК со следующей последовательностью:
[501] 5' - G G С C C C A U C G U C U A G C G G U U A G G A C G C G G C C C U C U C A A G G C C G A A A C G G G G G U U C G A U U C C C C U G G G G U C A C C A - 3' [576],
где 501 и 576 - номера первого и последнего нуклеотида.
В последовательности на 3'-конце триплет CCA, к которому присоединяется гуанин. координаты, где он присоединяется есть в pdb файле - Исследование вторичной структуры
С помощью программы find_pair пакета 3DNA были определены возможные водородные связи
между азотистыми основаниями 1n78.fp . В соответствии с полученными данными
Акцепторный стебель состоит из участка 501-507 и комплементарного ему участка 566-572.
* T-стебель состоит из участка 549-553 и комплементарного ему участка 561-565.
* D-стебель состоит из участка 510-513 и комплементарного ему участка 522-525.
* Антикодоновый стебель состоит из участка 539-543 и комплементарного ему участка 527-531.
скрипт для получения в RasMol изображения остова исследуемой тРНК, где акцепторный стебель выделен красным, Т-стебель - зеленым, D-стебель - синим, антикодоновый - оранжевым.
Структуру стеблевых дуплексов поддерживают 19 канонических и 2 неканонических пар оснований.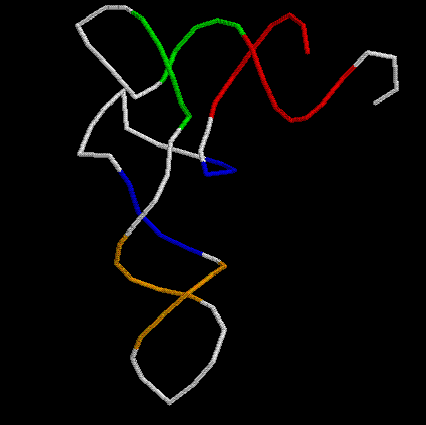 Рис.1. Вторичная структура глутаминовой тРНК из Thermus thermophilus
Рис.1. Вторичная структура глутаминовой тРНК из Thermus thermophilus
Скрипт для получения изображения define stem_acceptor 501-507,566-572 define stem_t 549-553, 561-565 define stem_d 510-513, 522-525 define stem_anticodon 539-543, 527-531 select all wireframe off cpk off backbone 100 select stem_acceptor color red select stem_t color green select stem_d color blue select stem_anticodon color orange select not (stem_acceptor, stem_t, stem_d, stem_anticodon) color white restrict *:C
изображение неканонической пары U-G 502-571 в акцепторном стебле.
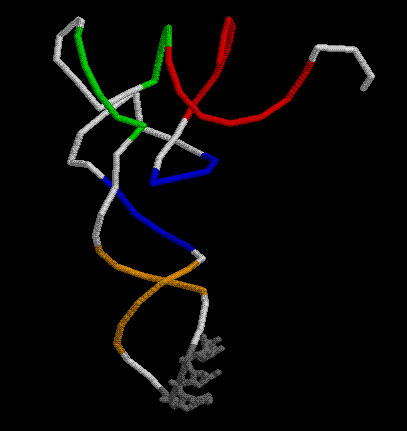
a) Вариабельная петля, видимо, отсутствует.
b) В Т-петле остатки тимидина отсутствуют.
c) В D-петле дигидроуридины отсутствуют.
*Дополнительное задание. антикодон СUC (534-536) обозначен серым цветом.
- Исследование третичной структуры
1. рассмотрим возможность стекинг-взаимодействия между
7 (0.004) C:.507_:[..A]Ax----U[..U]:.566_:C (0.006)
8 (0.004) C:.549_:[..G]G-----C[..C]:.565_:C (0.002)
принадлежащих концу акцепторного стебля и началу T стебля соответственно.
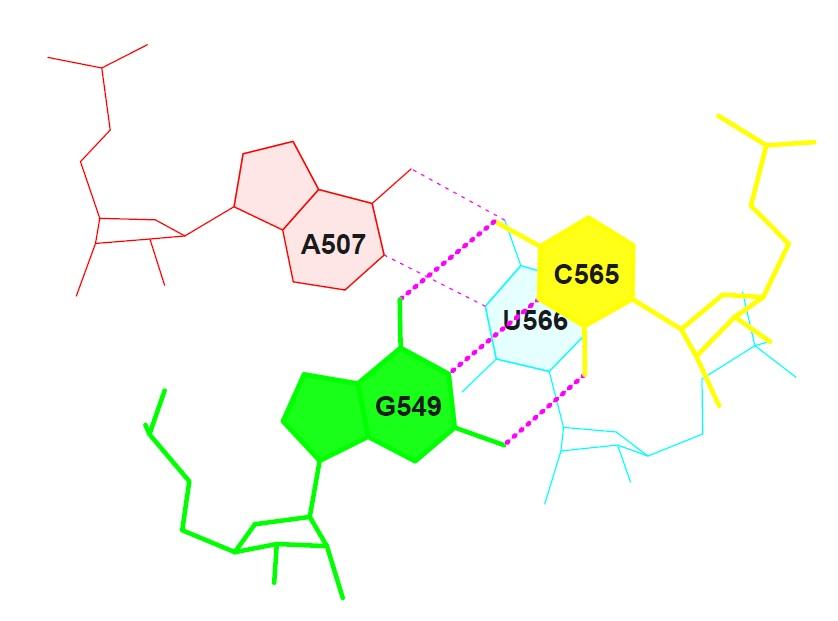
Две найденные пары не сильно перекрываются (всего 2.56 А), таким
образом вероятность появления стекинг-взаимодействия оценивается как низкая.
2. Дополнительная связь между основаниями D и T петель:
28 (0.004) C:.519_:[..G]Gx---xC[..C]:.556_:C (0.002)
- Предсказание вторичной структуры тРНК
Для предсказания вторичной структуры тРНК используем алгоритм Зукера.
Запустим программу mfold по последовательности.
При ее запуске можно изменять значение параметра P.
Этот параметр устанавливает, на сколько процентов выдаваемое предсказание
структуры может отличаться по своей вычисленной энергии от оптимального.
В моем случае при P=1 было получено вполне реальное изображение, а повышение P
дало только дополнительные варианты менее похожие на реальность:
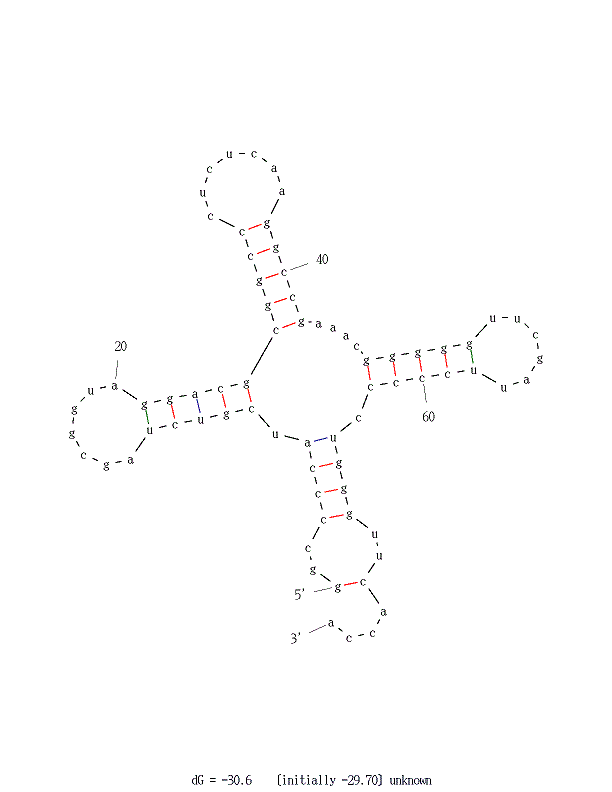
Участок структуры
(расшифровку названий см. на рис. 2 в статье О.О.Фаворовой)Позиции в структуре
(по результатам find_pair)Результаты предсказания
с помощью einvertedРезультаты предсказания
по алгоритму ЗукераАкцепторный стебель 5' 501-507 3'
5' 566-572 3'
Всего 7 парпредсказано 5 пар из 7 реальных предсказано 7 пар D-стебель 5' 510-513 3'
5' 522-525 3'
Всего 4 парыпредсказано 0 пар предсказано 4 пары и одна лишняя T-стебель 5' 549-553 3'
5' 561-565 3'
Всего 5 парпредсказано 0 пар предсказано 4 пары и одна лишняя Антикодоновый стебель 5' 538-544 3'
5' 526-532 3'
Всего 7 парпредсказано 5 пар предсказано 5 пар Общее число канонических пар нуклеотидов 19 предсказано 10 канонич. пар предсказано 17 канонич. пар
Программа einverted сумела частично предсказать акцепторный и антикодоновый стебли. При стандартных значениях параметров не было найдено ни одной пары, лишь уменьшение "Minimum score threshold" до "0" и параметра "gap penalty" до "4" привели к вышеуказанному результату. Дальнейшее снижение "gap penalty" приводило к огромному количеству лишних пар. Манипуляции с параметрами Match и Mismatch также не принесли ничего полезного.
из таблицы видно, что алгоритм Зукера более подходящий для определения структуры РНК,
чем einverted, которы больше подходит для ДНК, так как его резулятаты дают данные только
о комплементарных взаимодействиях и не зная самой структуры сложно понять где какой стебель.
© Garanina Irina