+-STRPN
+-4
+-3 +-STRP1
! !
! +---LACLM
!
--5 +STAES
! +--7
! +-6 +STAA1
! ! !
+-2 +--LISMO
!
! +-LACDA
+--1
+-LACAC
Вспомним, как выглядит правильное дерево
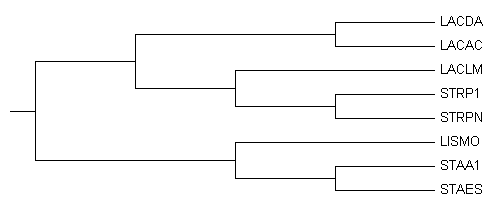
+-LACDA
+-LACDA !
! ! +---LACLM
! +STAES ! +-4
! +--6 ! ! ! +-STRP1
! +-5 +STAA1 ! ! +-3
! ! ! 2--5 +-STRPN
! ! +-LISMO ! !
1--4 ! ! +-LISMO
! ! +STRPN ! +-6
! ! +-3 ! ! +STAA1
! +-2 +STRP1 ! +--1
! ! ! +STAES
! +---LACLM !
! +-LACAC
+-LACAC
+---HSLU_LACAC
+----2
! +---HSLU_LACDA
+-----------------4
! ! +-------HSLU_LISMO
! +--3
! ! +-HSLU_STAA1
! +--------1
! +HSLU_STAES
!
! +-------------------------------------------------------FTSH_STRPN
! !
! ! +----CLPX_LACLM
5--6 +-10
! ! ! ! +-CLPX_STRP1
! ! ! +-9
! +----------11 +---CLPX_STRPN
! !
! ! +------CLPX_LISMO
! +-8
! ! +CLPX_STAA1
! +----7
! +CLPX_STAES
!
+----------------------------------------------CLPC_STAES
Два гомологичных белка будем называть ортологами, если они: