
Для отобранных на предыдущем занятии бактерий построили филогенетическое дерево, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Для этого сначала нашли последовательности 16S рибосомальной РНК каждой из бактерий. Это было сделано следующим способо: в записи EMBL, описывающей полный геном бактерии, нашли соответствующее поле (FT с FTkey rRNA с упоминанием 16S rRNA в примечании). Затем вырезали нужный участок из записи EMBL с использованием программы seqret.
| Название | Мнемоника | AC записи EMBL | Начало | Конец | Последовательность (+/-) |
| Clostridium tetani | CLOTE | AE015927 | 176113 | 177621 | + |
| Finegoldia magna | FINM2 | AP008971 | 611796 | 613319 | + |
| Lactobacillus delbrueckii | LACDA | CR954253 | 45160 | 46720 | + |
| Enterococcus faecalis | ENTFA | AE016830 | 248466 | 249987 | + |
| Lactococcus lactis | LACLM | AM406671 | 511423 | 512971 | + |
| Listeria monocytogenes | LISMO | AL591974 | 37466 | 39020 | + |
| Staphylococcus epidermidis | STAES | AE015929 | 1598006 | 1599559 | - |
Полученные описанным выше образом последовательности были сохранены в соответствующем файле, при этом названия последовательностей были отредактированы так, чтобы они отвечали мнемонике организмов. Выравнивание последовательностей было получено с помощью программы JalView (Web Service > Alignment > Muscle with Defaults).
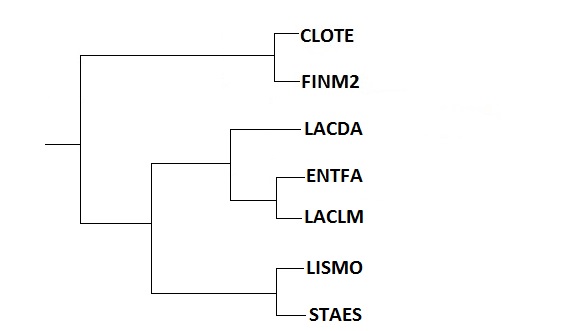
Затем выравнивание было импортировано в программу MEGA (как выравнивание нуклеотидных последовательностей), с помощью которой было построено дерево методом Neighbor-Joining. Полученное дерево приведено на изображении ниже.
|
|
| Neighbor-Joining | Исходное дерево |
Для гомологов белка CLPX_BACSU в отобранных бактериях необходимо построить дерево. Для поиска гомологов был предложен файл proteo.fasta, содержащий записи банка Uniprot, относящиеся к исходному списку бактерий. Поиск гомологов производился с помощью программы blastp гомологов (с порогом на E-value 0,001), а затем выбирали находки, относящиеся к отобранным бактериям. В файле gomolog.fasta получены последовательности гомологов CLPX_BACSU , относящиеся к отобранным бактериям. Затем построили выравнивание полученных последовательностей с помощью программы muscle. Полученный файл с выравниванием импортировали в программу MEGA, чтобы построить дерево. Ниже приведено дерево, реконструированное на основе выравнивания последовательностей гомологов белка CLPX_BACSU в отобранных бактериях методом Neighbor-Joining.
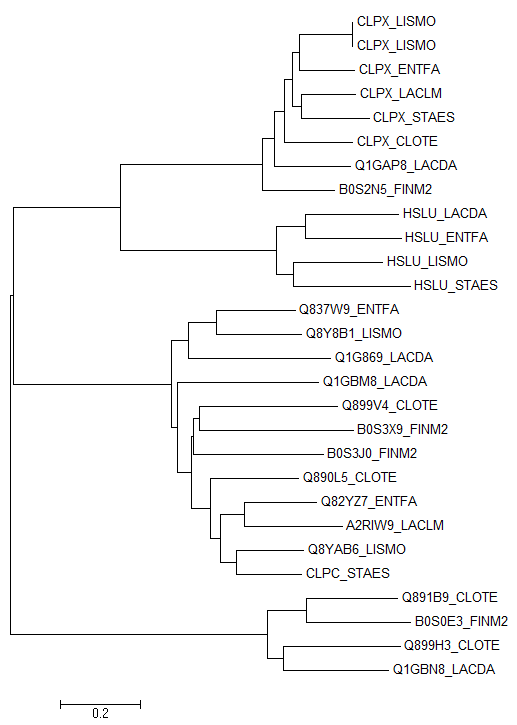 |
| Neighbor-Joining (CLPX_BACSU homologs) |
Если считать, что два гомологичных белка являются ортологами, и они из разных организмов, и разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования, то на построенном дереве ортологами являются, например, Q8YAB6_LISMO и CLPC_STAES, HSLU_LACDA и HSLU_ENTFA.
Если паралогами называть два гомологичных белка из одного организма, то из реконструированного дерева видно, что паралогами являются, например, B0S3X9_FINM2 и B0S3J0_FINM2, Q1G869_LACDA и Q1GBM8_LACDA.