� Пробные выравнивания
Определение положения фрагмента в полной последовательности
Для выполнения задания я запустил программу GeneDoc и импортировал в нее файл fellANDpart.fasta, содержащий последовательности фрагмента и всей полипептидной цепи белка.
Так выглядит фрагмент последовательности белка:
STAKLVKSKATNLLYTRNDVSD
А так выглядит последовательность всей полипептидной цепи белка:
MSTAKLVKSKATNLLYTRNDVSDSEKKATVELLNRQVIQFIDLSLKQAHWNMRGANFIAVHEMLDG
FRTALIDHLDTMAERAVQLGGVALGTTQVINSKTPLYPLDIHNVQDHLKELADRYAIVANDVRKAIGEAKDDDTADILTAASRDLDKFLWFIESNIE
После этого я двигал фрагмент последовательности белка, пока не добился полного совпадения букв.
Первый аминокислотный остаток фрагмента соответствует второму аминокислотному остатку полной последовательности белка, а последний - 23-му аминокислотному остатку полной последовательности.
Таким образом, заданный фрагмент соответствует позициям 2-23 в полной последовательности белка DPS_ECOLI.
Выравнивание было сохранено в файле alignment1.msf.
Затем была получена картинка с выравниванием. Она сохранена в файле aln1.gif:

Построение "наилучшего" выравнивания вручную
Я импортировал в GeneDoc файл shortseqs.fasta, содержащий два коротких фрагмента выравниваемых последовательностей.
Нужно выровнять последовательности, чтобы было сопоставлено максимальное число одинаковых букв при минимальном числе пропусков. Для этого будем оценивать вес выравнивания по следующей формуле:
W = M - nG,
где M - число совпавших букв, G - штраф за пропуск (равен 2), n - общее число пропусков.
Пропуском будем считать непрерывную последовательность символов "-" любой длины.
После выравнивания (сохраненного в файле alignment2.msf) была получена следующая картинка (сохраненная в файле aln2.gif):
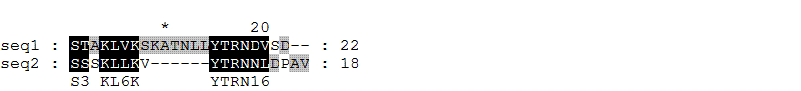
Итак, первая из последовательностей состоит из 22 аминокислотных остатков. вторая - из 18 аминокислотных остатков.
Длина выравнивания - 24.
Посчитаем теперь вес выравнивания по приведенной формуле: W = 8 - 2 . 1 = 6.
Наконец, посчитаем процент идентичности двух выровненных последовательностей (будем оценивать его как отношение числа колонок, в которых стоят одинаковые буквы, к общему числу колонок, включая "гэповые", умноженное на 100): 8 / 24 . 100% = 33.3%.
Поиск первой с N-конца выравнивания "близкородственной" замены а.о.
Для выполнения этого задания я воспользовался матрицей весов замен аминокислотных остатков BLOSUM62. Будем считать "близкородственными" заменами те, для которых значение элемента матрицы положительно. Тогда, как видно из выравнивания, первая "близкородственная" замена аминокислотных остатков встречается на второй позиции выравнивания. Это замена T на S (треонина (Thr) на серин (Ser)). Вес такой замены в соответствии с матрицей составляет 1.
"Близкородственность" двух этих аминокислотных остатков объясняется очень просто. Треонин и серин представляют собой очень похожие аминокислоты, отличающиеся всего на одну метильную группу, то есть являющиеся гомологами. Боковые группы этих аминокислот полярны (заканчиваются -OH группой). В общем, два этих аминокислотных остатка обладают одинаковыми свойствами, поэтому имеют положительное значение элемента матрицы. Однако вес этой замены не такой уж и большой, составляет всего 1. Это объясняется, прежде всего, тем, что, обладая гидроксильной группой, как и серин, треонин является слабо гидрофобным аминокислотным остатком, ведь засчет содержания метильной группы, атом углерода становится асcимметричным, а это затрудняет вращение гидроксильной группы (ведь крутиться она будет вместе с метильной). Поэтому свойства, пусть и похожи, но во многом отличаются. Впрочем, несмотря на небольшой вес, замещение одного остатка на другой в процессе эволюции весьма и весьма вероятно.
Дополнительные задания
Определение % сходства выровненных фрагментов
Теперь посчитаем процент сходства выравненных фрагментов, опираясь на матрицу весов замен а.о. BLOSUM62. Процентом сходства будем считать отношение числа колонок со сходными буквами к общему числу, умноженное на 100%. Сходными буквами же будем считать такие, для которых значение элемента BLOSUM62 положительно.
Для этого обратимся еще раз к выравненным последовательностям:
Помимо 8 совпавших букв имеем 8 пар букв. Рассмотрим эти пары.
Первая из них (расположенная на позиции 2) - пара T-S (треонин (Thr) и серин (Ser)). Значение элемента матрицы для этой пары - +1. Следовательно, эти аминокислотные остатки сходны (подробнее их сходство рассмотрено в обязательном задании 3).
Вторая из них (расположенная на позиции 3) - пара A-S (аланин (Ala) и серин (Ser)). Значение элемента матрицы для этой пары - +1. Следовательно, эти аминокислотные остатки сходны (и правда, боковые группы аминокислотных остатков отличаются только на одну гидроксильную группу).
Третья из них (расположенная на позиции 6) - пара V-L (валин (Val) и лейцин (Leu)). Значение элемента матрицы для этой пары - +1. Следовательно, эти аминокислотные остатки сходны (дейстивтельно, боковые группы обоих этих аминокислотных остатков алифатические, а значит гидрофобные, имеющие довольно длинные хвосты; кроме того, отличаются они всего на одну -CH2 группу, то есть являются гомологами, а значит, обладают сходными свойствами).
Четвертая из них (расположенная на позиции 8) - пара S-V (серин (Ser) и валин (Val)). Значение элемента матрицы для этой пары - -2. Следовательно, эти аминокислотные остатки не сходны (действительно, боковая группа серина меньше, кроме того, она полярна, в отличие от боковой группы валина, которая является гидрофобной и более громоздкой).
Пятая из них (расположенная на позиции 19) - пара D-N (аспарагиновая кислота (Asp) и аспарагин (Asn)). Значение элемента матрицы для этой пары - +1. Следовательно, эти аминокислотные остатки сходны (и правда, боковые группы аминокислотных остатков отличаются только тем, что карбоксильная группа аспарагиновой кислоты в боковой группе заменена у аспарагина на амидную).
Шестая из них (расположенная на позиции 20) - пара V-L (валин (Val) и лейцин (Leu)). Значение элемента матрицы для этой пары - +1. Следовательно, эти аминокислотные остатки сходны (сходство этих 2 аминокислотных остатков уже было рассмотрено).
Седьмая из них (расположенная на позиции 21) - пара S-D (серин (Ser) и аспарагиновая кислота (Asp)). Значение элемента матрицы для этой пары - 0. Следовательно, эти аминокислотные остатки не сходны (действительно, боковая группа аспарагиновой кислоты имеет карбоксильную группу, то есть является кислотой, а значит, заряженным аминокислотным остатком, в отличие от серина, у которого на том же месте находится гидроксильная группа, которая делает аминокислотный остаток полярным, но не заряженным).
Ну и, наконец, последняя из них (расположенная на позиции 22) - пара D-P (аспарагиновая кислота (Asp) и пролин (Pro)). Значение элемента матрицы для этой пары - -1. Следовательно, эти аминокислотные остатки не сходны (и правда, они совершенно не сходны, ведь пролин вообще является иминокислотой, то есть его боковая группа образует цикл с N-концом аминокислоты, аспарагиновая же кислота является заряженной кислотой).
Таким образом, получаем 5 пар сходных букв помимо 8 совпавших. Тогда процент сходства выровненных фрагментов становится равным: (8 + 5) / 24 . 100% = 54.17%.
Построение иного выравнивания двух заданных фрагментов с весом не хуже, чем получилось в обязательном упр.2
Смотря на выравнивание, сделанное в первом задании, я думал, что сложно уже что-то исправить, чтобы выравнивание стало еще лучше. Однако я решил переставить в выравнивании всего одну букву на другое место и получил выравнивание, в котором сходна стала еще одна пара букв. Это выравнивание было сохранено мной в файле alignment3.msf, а его картинка сохранена в файле aln3.gif. Вот так выглядит это выравнивание:
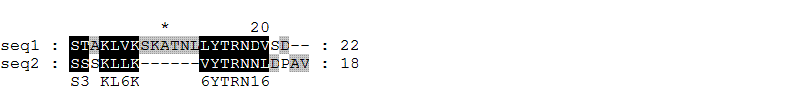
Как видно из рисунка, теперь валин во втором фрагменте расположен не под серином, с которым он явно не был сходен, а под лейцином, свойства которого очень похожи на свойства валина (обе эти аминокислоты являются громоздкими и гидрофобными, их боковые группы отличаются всего на одну -CH2 группу, то есть являются гомологами). В матрице замен а.о. значение элемента для этой пары равно +1.
Таким образом, для такого выравнивания процент идентичности остался тем же (33.3%).
А процент сходства для этого выравнивания равен: (8+6) / 24 . 100% = 58.33%.
Получение с помощью функций Excel из матрицы Blosum списка замен вида "W-Y вес=..."
Чтобы составить таблицу, которая сможет считать вес каждой пары букв в данном выравнивании, вначале я вставил в лист Excel матрицу замен а.о. BLOSUM62.
Затем на другом листе в ячейках A1 и A2 я записал последовательности букв моего выравнивания (вместе с пробелами), под ними (в ячейках B3 и B4) формулу, которая позволит посчитать число букв с пробелами в каждом из фрагментов, а затем в ячейке B5 - формулу, которая позволит посчитать длину выравнивания (которая будет равна максимальной из длин двух участвующих в выравнивании фрагментов). Далее помещена таблица, в ячейках которой записаны формулы, показывающие буквы фрагментов в столбик в отдельных ячейках, и, собственно, вес каждой пары букв из двух фрагментов (с помощью функций ВПР и ПОИСКПОЗ).
Таким образом, чтобы посчитать вес каждой пары букв в выравнивании, требуется просто ввести в ячейки A1 и A2 последовательности букв фрагментов, участвующих в выравнивании. Остальное таблица посчитает сама.
Кроме того, таблица считает вес всего выравнивания. Для этого она суммирует вес каждой пары букв в выравнивании, но не учитывает вес каждого пробела в пропуске (учитывает вес всего пропуска как вес одного пробела, то есть -4), а так же пробелы, полученные по причине конца последовательности одного из фрагментов. Суммарный вес выравнивания можно посмотреть под ячейкой SUMweight справа от весов каждой пары букв.
Однако для того, чтобы воспользоваться этой функцией (подсчета суммарного веса), нужно в конце последовательности букв каждого фрагмента ввести столько пробелов, сколько не хватает для заполнения в столбик всех 40 ячеек. Тогда все будет работать=)
Вот ссылка на таблицу: weight.xls.
Назад