|
 |
Учебный сайт Морозова Александра |
<< Назад к странице 4 семестра
Коллоквиум 1Из практикума 3:Задания:1) Реконструируйте методом максимальной экономии укоренённое дерево отобранных вами бактерий, используя в качестве внешней группы белок того же семейства из сенной палочки (Bacillus subtilis, мнемоника BACSU в Uniprot); 2) Проведите бутстрэп-анализ филогении своих белков, используя один из методов, доступных из программы MEGA; 3) Постройте филогенетическое дерево тех же бактерий, что в предыдущих заданиях, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Результаты
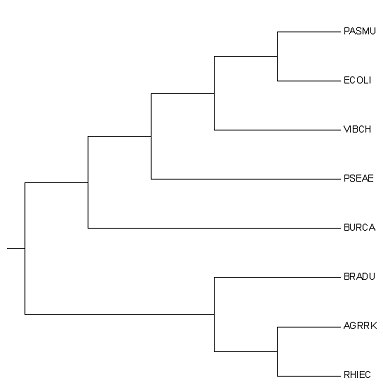
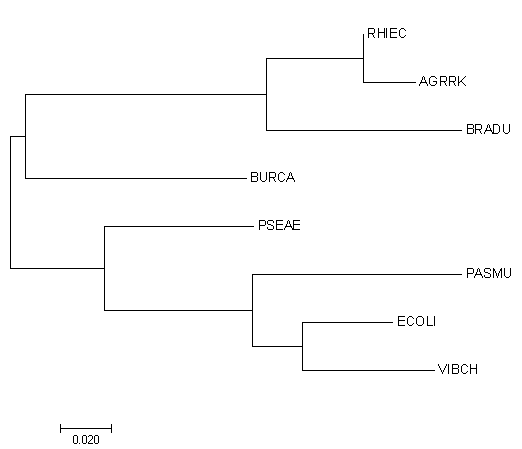
1. Укоренение с использованием внешней группы.1) Получил последовательности белков из отобранных бактерий и BACSU путем использования формы загрузки из Uniprot. Использовал тот же файл с идентификаторами, что и в практикуме 2, с добавлением идентификатора BACSU. 2) Командой: muscle -in efts_bacsu.fasta -out bacsu_efts_alignment.fasta получил выравнивание белков, которое затем обработал в программе Jalview. Отредактированное выравнивание доступно по ссылке. 3) Загрузив выравнивание в MEGA, реконструировал дерево методом Maximum parsimony. Далее по схеме Sub Tree -> Root, выбрав тривиальную ветвь BACSU, укоренил дерево. Затем, нажав на значок Show Subtree Separately и вновь выбрав тривиальную ветвь BACSU, получил изображение укорененного дерева без BACSU. Изображение дерева см. ниже. Дерево сохранил в формате Newick в файл root_efts_tree.tre
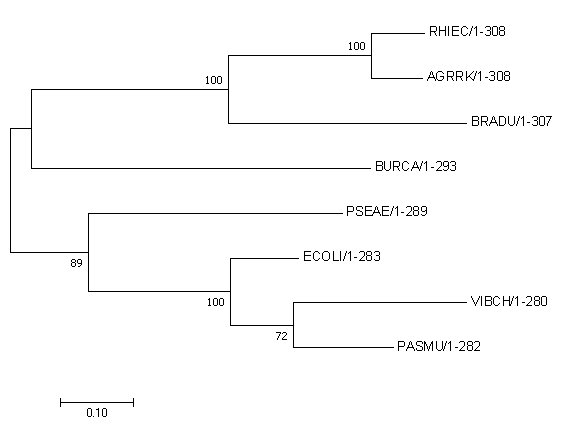
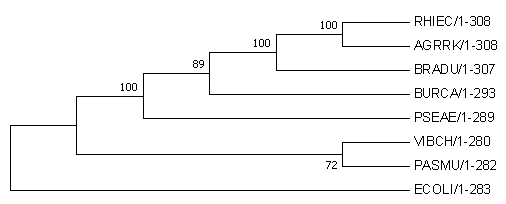
2. Бутстрэп.1) Для реконструкции использовал метод Наибольшего правдоподобия. В результате выполнения программы получил 2 неукорененных дерева: Original tree и Bootstrap consensus tree, с указанием на ветвях значений бутстрэп-поддержки. Ниже представлены изображения полученных деревьев. Деревья в Newick-формате доступны по ссылкам: Original tree Bootstrap consensus tree
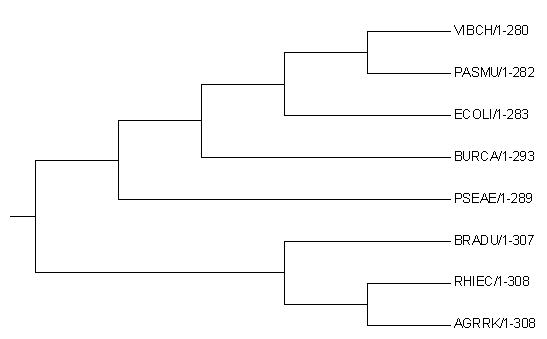
3. Построение дерева по нуклеотидным последовательностям.1) Обнаружил, что все последовательности, кодирующие РНК (не только 16s рРНК) данного организма расположены в файле с расширением .frn. Поиск 16s рРНК в файле осуществлял с помощью Ctrl-F, запись в fasta-файл с помощью простого копирования и вставки. В среднем в файлах были найдены от 6 до 8 последовательностей, кодирующих 16s рРНК. В процессе поиска геномов организмов в списке для некоторых из них пришлось использовать синонимичные названия, представленные в NCBI Taxonomy Browser. 2) Выравнивание построено командой: muscle -in 16s_rrna.fasta -out 16s_rrna_alignment.fasta Выравнивание доступно по ссылке. 3) Выравнивание было импортироано в программу MEGA. Филогенетическое дерево было реконструировано методом Наибольшего правдоподобия. Результат, сохраненный в формате Newick, доступен по ссылке. Изображение полученного дерева представлено ниже.
|
||||||||||