
| AC | ID | Функция домена | Ссылка на страницу домена в Pfam |
| PF14759 | Reductase_С | Reductase C-terminal | http://pfam.xfam.org/family/PF14759 |
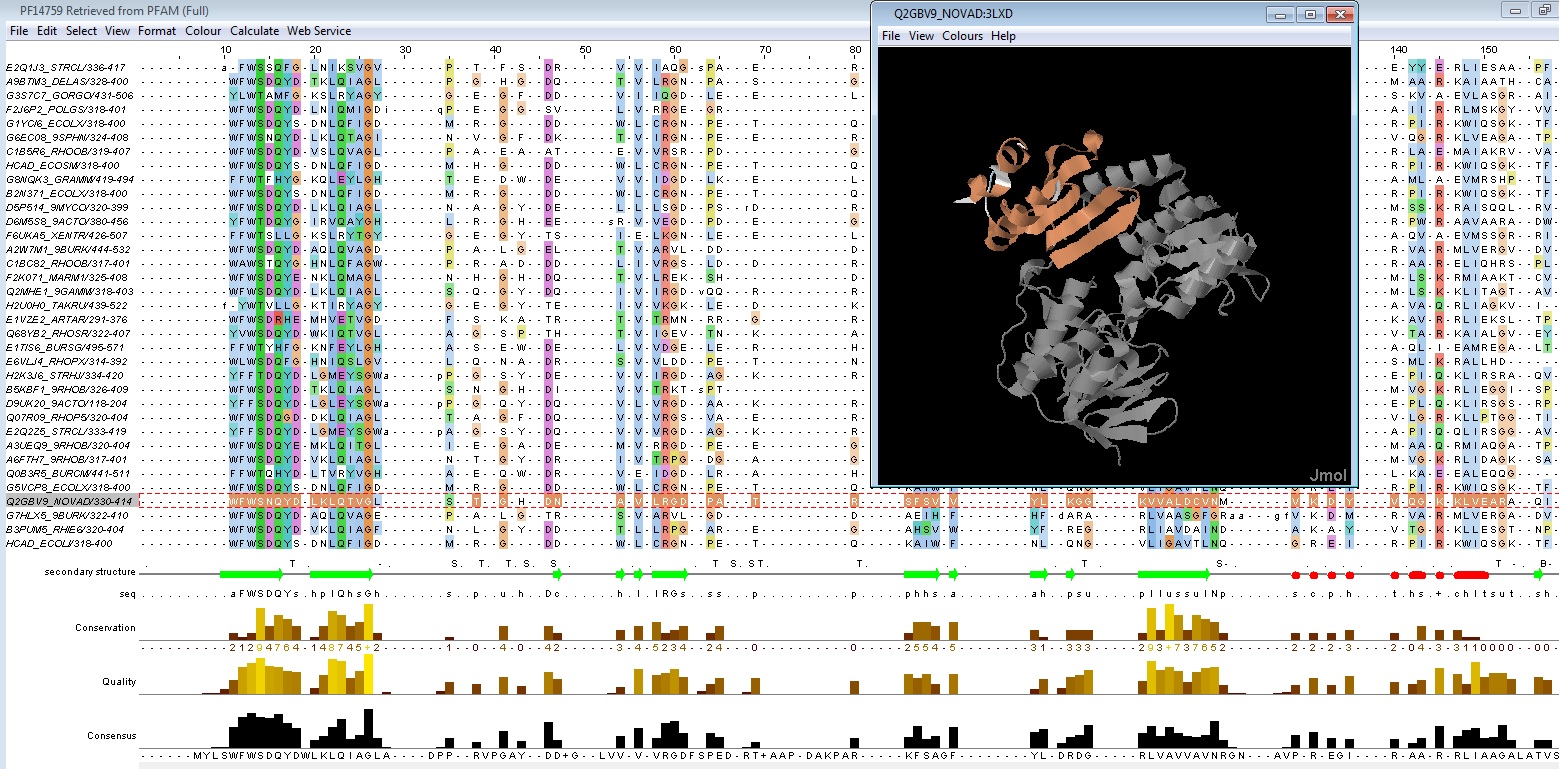

 Ссылка на выравнивание в формате jar
✧ Вывод: одну последовательность пришлось вырезать, так как она плохо выравнивалась (A1L230_DANRE). На выравнивание можно четко увидеть домен Reductase_C, входящий в обе архитектуры, причем этот домен присутствует у архей, у бактерий и у эукариот. Домен на выравнивании представлен правильно, поскольку в качестве концов выбиралась последовательность, в которой по Pfam были круглые концы.
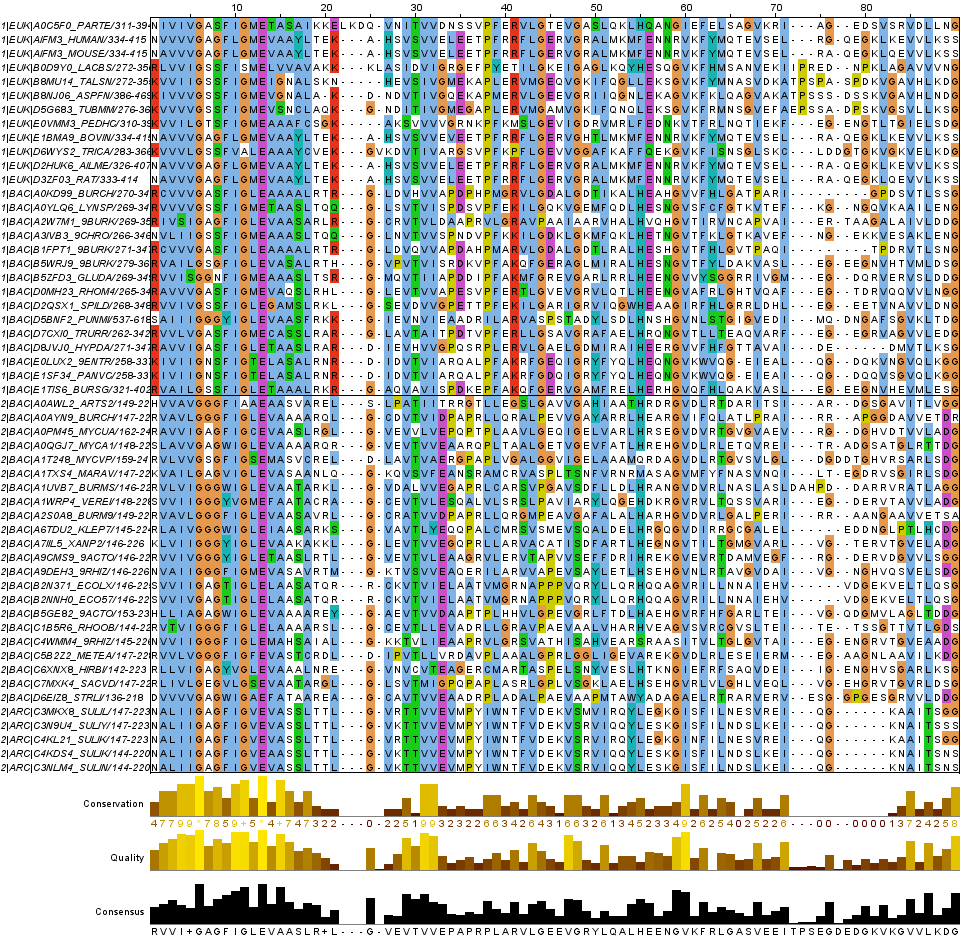
Ссылка на выравнивание в формате jar
✧ Вывод: одну последовательность пришлось вырезать, так как она плохо выравнивалась (A1L230_DANRE). На выравнивание можно четко увидеть домен Reductase_C, входящий в обе архитектуры, причем этот домен присутствует у архей, у бактерий и у эукариот. Домен на выравнивании представлен правильно, поскольку в качестве концов выбиралась последовательность, в которой по Pfam были круглые концы.
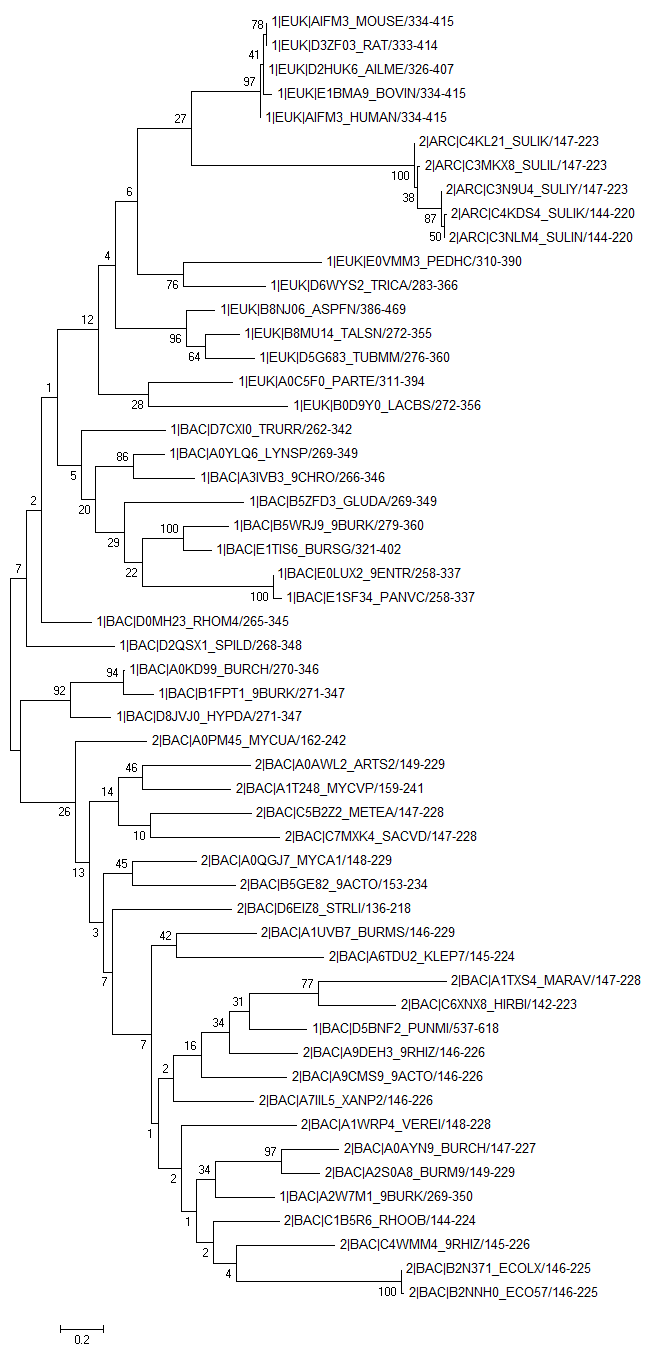 2) Bootstrap consensus tree (редактированное и укорененное)
2) Bootstrap consensus tree (редактированное и укорененное)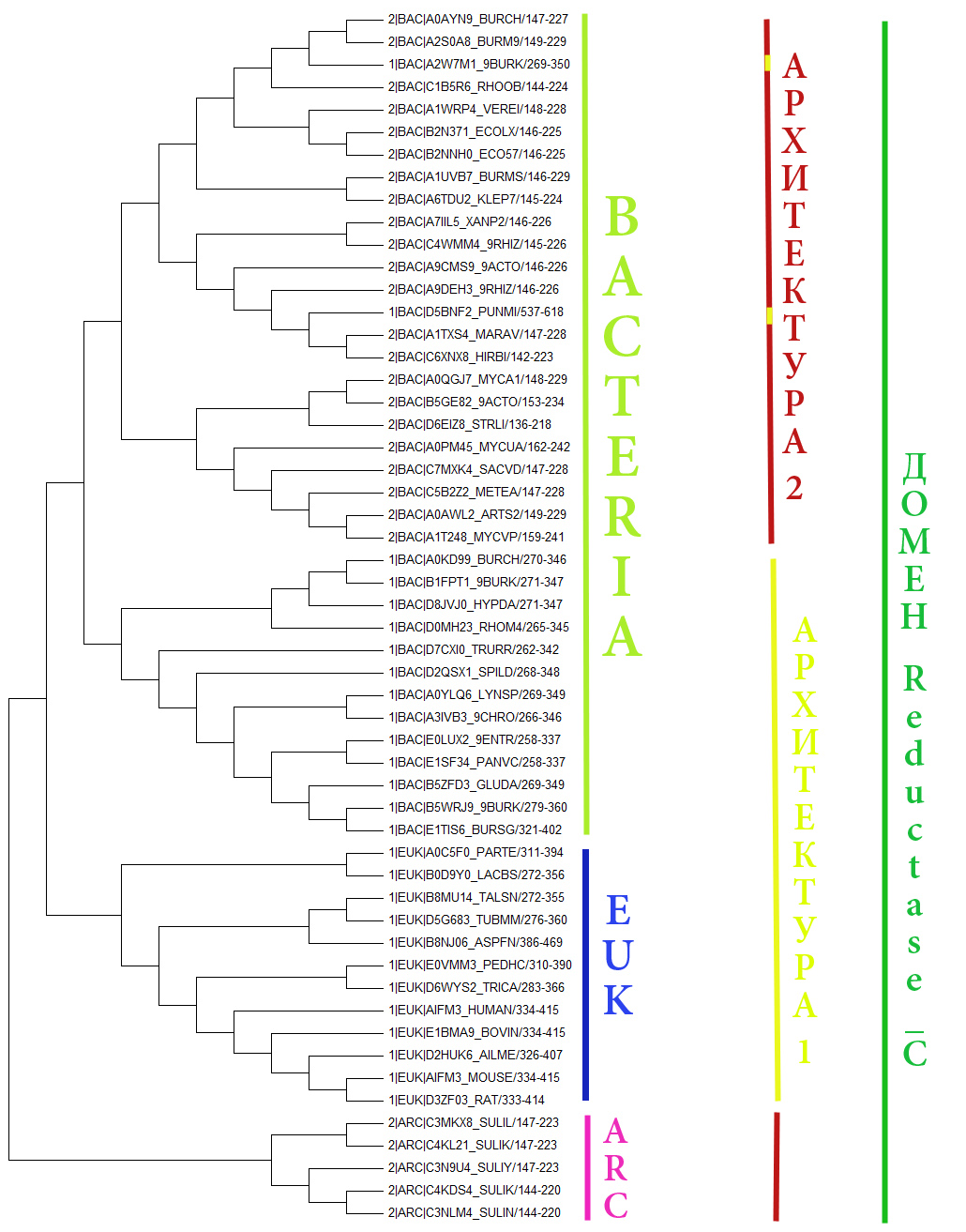 Вывод: структура дерева правильная. Причем, методом проб и ошибок, было проверено, что данное дерево по одному конкретному домену в двух архитектурах практически ничем не отличается от дерева, построенного по полным белковым последовательностям. Слабую поддержку имеет ветвь, отделяющая архей столь неправдоподобно, с чем я согласен. Однако на редактированном bootstrap consensus tree укоренил как раз в архей, получилось дерево, которое я считаю наиболее правильным, поскольку общий предок имел более простую архитектуру, однако и эукариот и у бактерий на С-конце появляется новый домен Rieske, что значительно усложняет архитектуру, а следовательно, появились новые функции белка. Независимые делеции С-концевого участка четко видно на примере A2W7M1_9BURK и D5BFN2_PUMNI. На original tree можно увидеть, что ветки отделяющие этих представителей имеют слабую поддержку.
Вывод: структура дерева правильная. Причем, методом проб и ошибок, было проверено, что данное дерево по одному конкретному домену в двух архитектурах практически ничем не отличается от дерева, построенного по полным белковым последовательностям. Слабую поддержку имеет ветвь, отделяющая архей столь неправдоподобно, с чем я согласен. Однако на редактированном bootstrap consensus tree укоренил как раз в архей, получилось дерево, которое я считаю наиболее правильным, поскольку общий предок имел более простую архитектуру, однако и эукариот и у бактерий на С-конце появляется новый домен Rieske, что значительно усложняет архитектуру, а следовательно, появились новые функции белка. Независимые делеции С-концевого участка четко видно на примере A2W7M1_9BURK и D5BFN2_PUMNI. На original tree можно увидеть, что ветки отделяющие этих представителей имеют слабую поддержку.