Практикум 8: Нуклеотидный BLAST

Поиск гена, кодирующего δ-субъединицу АТФ-синтазы
Поиск начался с фразы "ATP synthase subunit delta", а потом пошло поехало...
- Идентификатор белка - XP_004285918.1, его последовательность
- Идентификатор нуклеотидной записи, к которой относится ген, кодирующий данный белок - NC_064561.1 (698461..700524), Gene ID: 101290442. Расположение: 3 хромосома.
- Последовательность небольшой окрестности этого гена.
Локализация и идентификация гена:
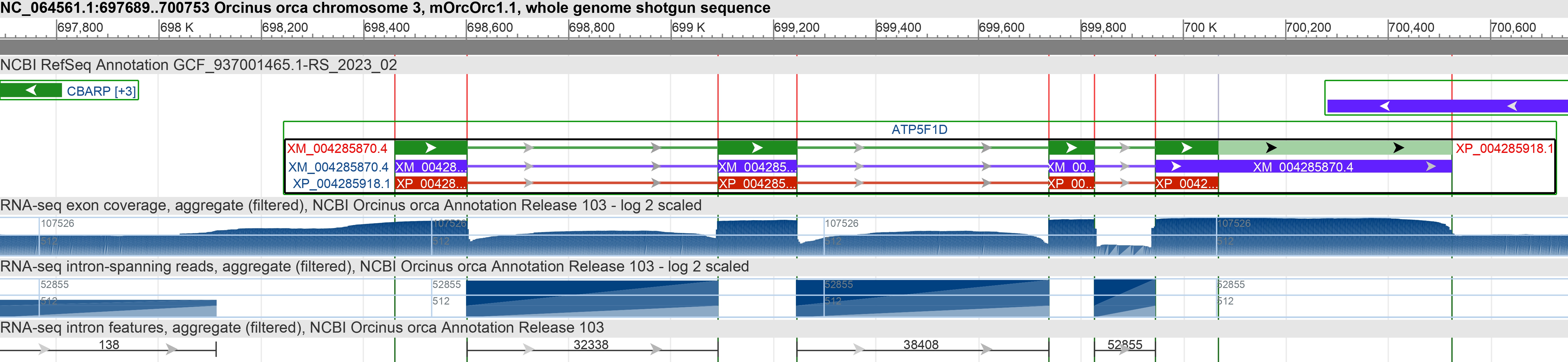
- Ген дельта-субъединицы АТФ-синтазы (ATP5F1D) расположен на прямой цепи хромосомы 3 (координаты ~697,689 – 700,753 н.). Для его поиска в геномном браузере использовался идентификатор белка XP_004288918.1.
Особенности структуры:
- На схеме ген ATP5F1D показан зелёным. Интересно, что он имеет два транскрипционных варианта (фиолетовые блоки), которые дают начало двум разным кодирующим последовательностям (CDS, красные). Однако эти последовательности кодируют идентичную белковую последовательность (XP_004288918.1). Направление считывания гена указано стрелками.
Геномное окружение:
- Слева от ATP5F1D (верх по потоку) на обратной цепи расположен ген CBARP, а справа (вниз по потоку) — ген C1QL4, также на обратной цепи.
Экспрессионное подтверждение:
- Наличие и корректность аннотации гена ATP5F1D подтверждается представленными внизу рисунка данными RNA-seq, демонстрирующими его активную экспрессию.
Разные варианты BLAST для фрагмента ДНК
Исходя из таксономической удалённости, для поиска гомологов было выбрано семейство Пауки (Araneae), относящееся к первичноротым (Protostomia), в то время как исходный организм (косатка) является вторичноротым (Deuterostomia). Эволюционная дистанция между этими группами превышает 500 млн лет, что позволяет оценить консервативность гена ATP5F1D.
Поиск проводился в базе данных RefSeq Genome Database (refseq_genomes), которая на момент анализа содержала 4 сборки геномов пауков (значение указано в поле "Database" на странице результатов BLAST). Для сравнения, поиск в NCBI Datasets показывает, что в таксоне Araneae имеется больше сборок, но в референсную базу включены только аннотированные и курируемые геномы.
Алгоритм blastn
Алгоритм blastn был использован для прямого сопоставления нуклеотидных последовательностей. В качестве запроса выступила кодирующая область (CDS) гена ATP5F1D, взятая из транскрипта XM_004288970.4 (длина 507 п.н.). Поиск проводился в базе референсных геномов (refseq_genomes) с ограничением по таксону Araneae. Из доступных вариантов был выбран режим blastn (Optimize for somewhat similar sequences), поскольку он, в отличие от megablast, рассчитан на обнаружение дивергентных последовательностей и не требует высокой степени идентичности (>95%).
В результате значимых совпадений обнаружено не было. Это указывает на то, что нуклеотидная последовательность гена у пауков значительно дивергировала и не сохранила достаточного сходства для детекции методом прямого сравнения ДНК.
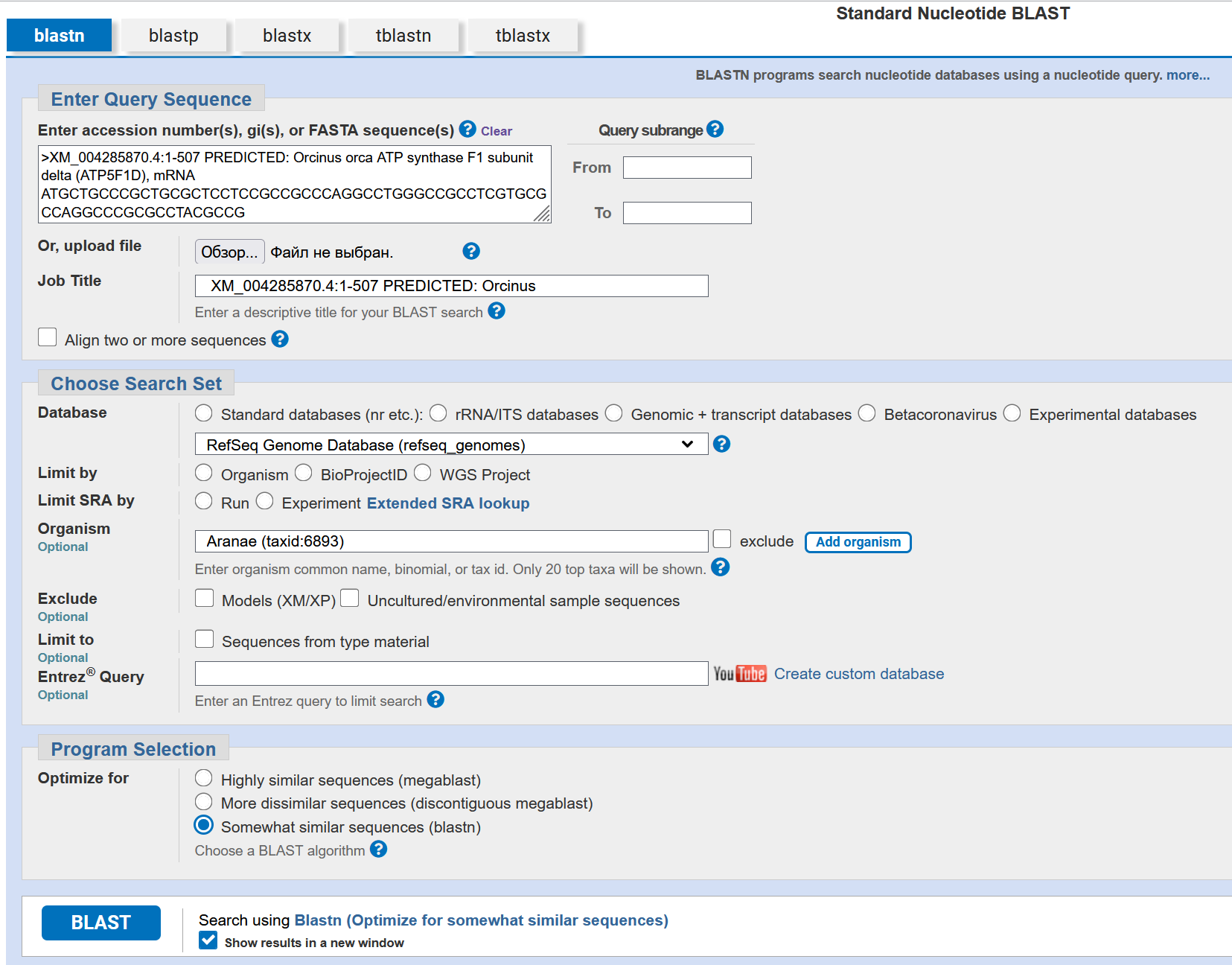
Алгоритм tblastn
Алгоритм tblastn был применён для поиска на уровне аминокислотных последовательностей. Запросом служила последовательность белка δ-субъединицы АТФ-синтазы косатки (XP_004288918.1, 168 а.к.). База данных и таксономическое ограничение остались прежними. Алгоритм tblastn транслирует все шесть рамок считывания нуклеотидных последовательностей в базе данных и сравнивает их с белковым запросом, что позволяет выявить консервативные участки даже при высокой дивергенции ДНК.
В отличие от blastn, tblastn обнаружил 4 значимых совпадения — по одному в каждом из четырёх геномов пауков. Найденные гомологи принадлежат видам Stegodyphus dumicola (идентичность 62,75%, e-value 6e-14), Uloborus diversus (60,78%, 2e-13), Parasteatoda tepidariorum (53,85%, 3e-12) и Argiope bruennichi (57,14%, 9e-11). Высокие статистические показатели и идентичность свыше 50% подтверждают, что это ортологичные гены, кодирующие консервативную δ-субъединицу АТФ-синтазы.
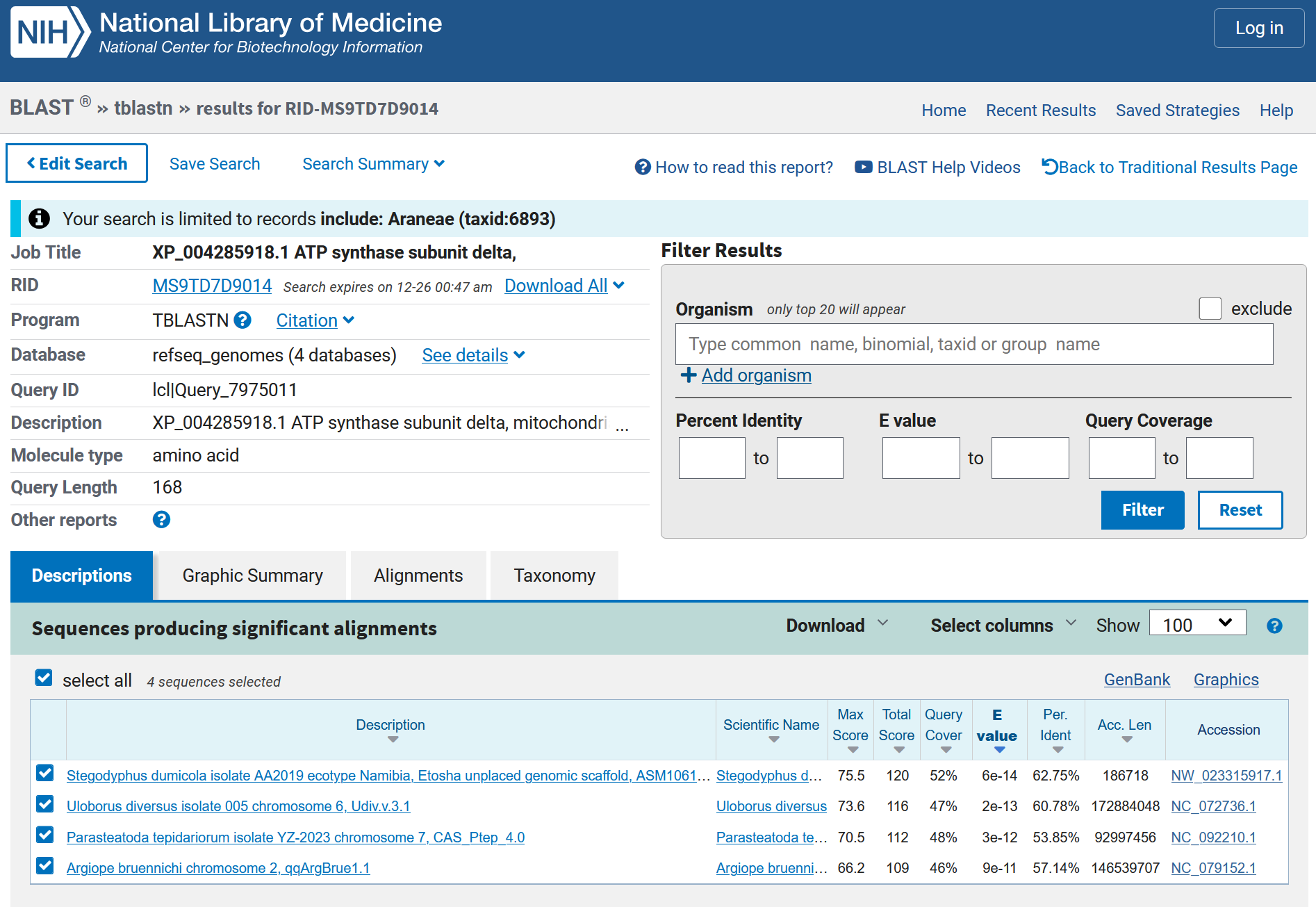
Таким образом, прямое сравнение нуклеотидных последовательностей (blastn) не дало результатов, в то время как поиск с учётом аминокислотной последовательности (tblastn) успешно выявил гомологи во всех доступных геномах. Это наглядно демонстрирует разную скорость эволюции ДНК и белка: нуклеотидная последовательность за более чем 550 млн лет дивергенции накопила слишком много замен для детекции методом blastn, тогда как аминокислотная последовательность сохранила консервативные черты, позволившие tblastn идентифицировать ортологи.
Гены основных рибосомальных РНК по далекому гомологу
Для поиска генов рибосомальных РНК (рРНК) был использован метод сравнения с эволюционно далёким гомологом. В качестве мишени выступил митохондриальный геном косатки (Orcinus orca), а в качестве запроса — последовательности бактериальных рРНК кишечной палочки (Escherichia coli). Такой подход оправдан эндосимбиотическим происхождением митохондрий: их рибосомы сохранили гомологию с бактериальными.
Подготовка данных и локальный поиск BLAST
Работа выполнялась на сервере в терминальной среде. Сначала последовательность митохондриального генома косатки (ID: NC_064558.1, длина 16 392 п.н.) была проиндексирована для локального поиска с помощью утилиты makeblastdb:
makeblastdb -in orca_mitochondrial.fasta -dbtype nucl -out orca_mito_db -title "Orcinus orca mitochondrial genome"
Запросы были представлены в виде файла rRNA_ecoli.txt, содержащего последовательности 16S и 23S рРНК E. coli в формате multi-FASTA. Для удобства файл был автоматически разделен на два: 16S_rRNA.fasta и 23S_rRNA.fasta. Поиск гомологов проводился отдельно для каждого типа рРНК с использованием алгоритма blastn. Учитывая значительное эволюционное расстояние, параметры поиска были ослаблены для повышения чувствительности: увеличен порог e-value до 0.1, уменьшена длина слова (word size) до 7 и отключён фильтр низкокомплексных последовательностей (-dust no).
blastn -query 16S_rRNA.fasta -db orca_mito_db -out results_16S.txt -outfmt 7 -evalue 0.1 -word_size 7 -dust noblastn -query 23S_rRNA.fasta -db orca_mito_db -out results_23S.txt -outfmt 7 -evalue 0.1 -word_size 7 -dust no
Результаты поиска и их интерпретация
Для 16S рРНК E. coli было обнаружено три значимых совпадения в митохондриальном геноме. Все они представляют собой короткие, но высококонсервативные участки (идентичность 87.5-100%), расположенные в области ~310 – 1018 нуклеотидов (наиболее значимое: e-value = 0.004, идентичность 100% на участке 17 п.н.). Полные результаты сохранены в файле results_16S.txt.
Для 23S рРНК E. coli было обнаружено четыре значимых совпадения, сгруппированных в области ~1400 – 2532 нуклеотидов. Наиболее значимое из них имеет e-value = 1.67e-04 при идентичности 86.111% на участке длиной 36 п.н. Результаты сохранены в файле: results_23S.txt
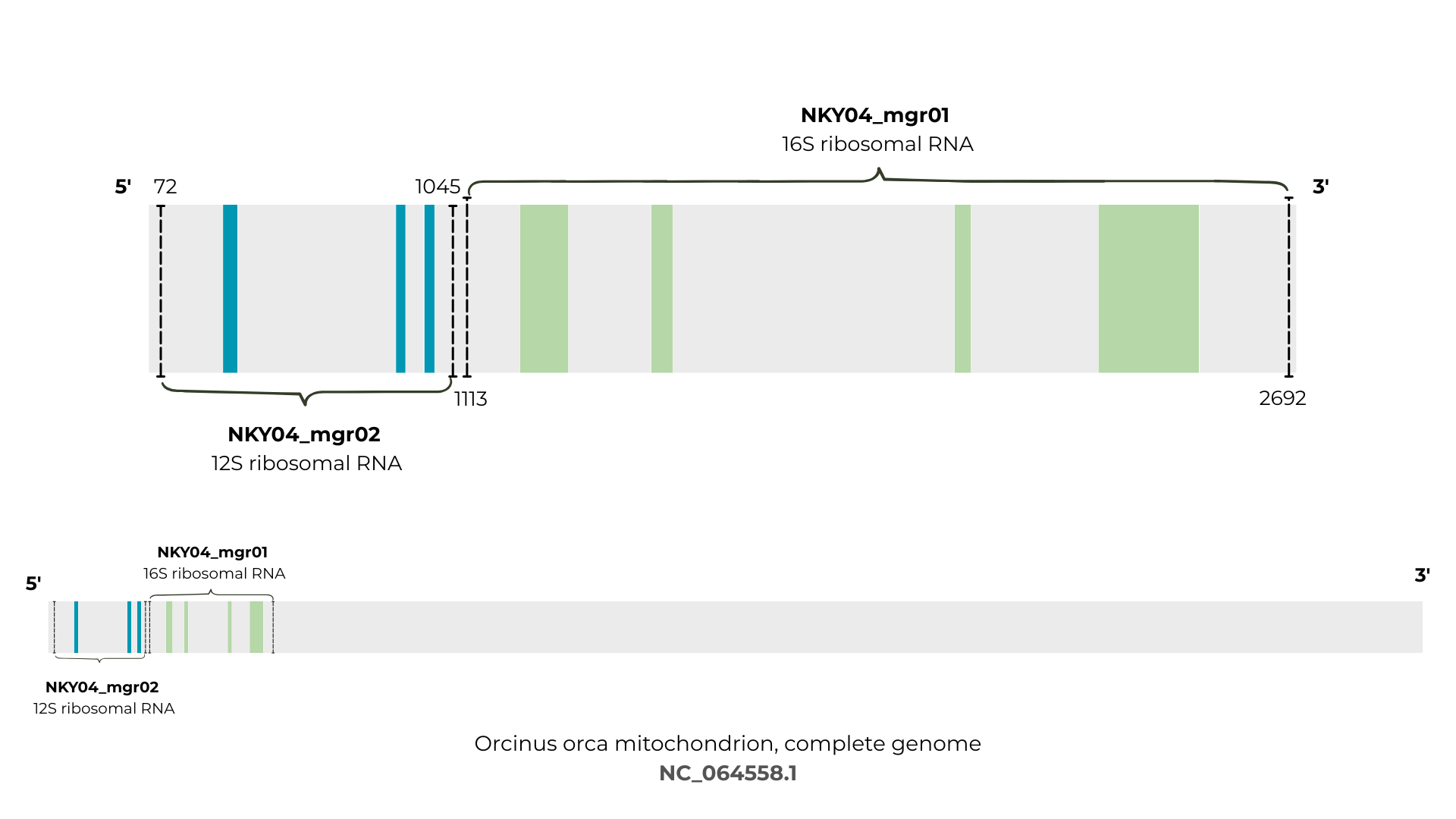
Успех поиска по отдалённому гомологу. Несмотря на использование последовательностей из другого домена жизни (бактерии), локальный BLAST успешно выявил гомологичные участки в митохондриальном геноме эукариота. Это подтверждает универсальную консервативность ключевых функциональных элементов рибосом.
Идентификация генов. Пространственная группировка разрозненных BLAST-совпадений в два неперекрывающихся кластера позволила идентифицировать их как известные гены митохондриальных рРНК млекопитающих: 12S рРНК и 16S рРНК.
Эволюционное подтверждение. Сам факт обнаружения гомологии является молекулярным свидетельством в пользу эндосимбиотической теории происхождения митохондрий. Результаты показывают, что за миллиарды лет эволюции общая нуклеотидная последовательность генов рРНК сильно дивергировала, однако отдельные, вероятно, критически важные для сборки или функции рибосомы, мотивы остались узнаваемыми.
Карты локального сходства
Для анализа были выбраны два вируса из рода Morbillivirus, поражающие разных хозяев:
- Phocine distemper virus (вирус чумы тюленей), штамм PDV/Wadden_Sea.NLD/1988. Идентификатор RefSeq: NC_028249.1. Длина генома: 15 696 п.н.
- Dolphin morbillivirus (морбилливирус дельфинов). Идентификатор RefSeq: NC_005283.1. Длина генома: 15 702 п.н.
Поиск был выполнен на сайте NCBI с помощью Nucleotide Advanced Search. Использовались фильтры: Organism: "Morbillivirus", Title: "complete genome". В результатах были отобраны только записи из курированной базы RefSeq.
Данные вирусы (Phocine distemper virus и Dolphin morbillivirus) выбраны как патогены морских млекопитающих (ластоногих и китообразных), что тематически продолжает предыдущую работу с косаткой. Их принадлежность к одному роду (Morbillivirus) обеспечивает общее геномное строение, а адаптация к разным хозяевам создаёт необходимый уровень нуклеотидной дивергенции для наглядного сравнения алгоритмов Megablast и BLASTN.
Карты локального сходства (Dot Plots) строились с помощью онлайн-инструмента BLAST 2 Sequences на портале NCBI. Для одной и той же пары последовательностей было выполнено четыре запуска с разными алгоритмами и параметрами:
- 1. Megablast с параметром word size = 24 (ослабленный режим).
- 2. Megablast с параметром word size = 16 (чувствительный режим).
- 3. BLASTN (Somewhat similar sequences) со стандартными параметрами.
- 4. TBLASTX со стандартными параметрами.
Результаты и анализ карт локального сходства
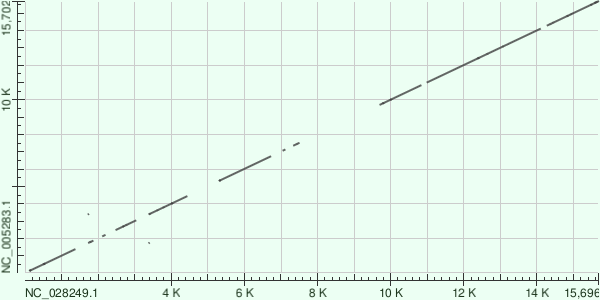
Карта Megablast (word size = 24)
На карте видна лишь одна короткая диагональная черта. Это означает, что при такой длине "слова" (24 нуклеотида) алгоритм смог обнаружить только единственный наиболее консервативный участок генома. Остальные регионы имеют уровень дивергенции выше порога чувствительности для этого строгого режима.
Карта Megablast (word size = 16)
Уменьшение параметра word size до 16 повысило чувствительность. На карте проявилась длинная, но чётко прерывистая диагональ. Скорее всего, её сегменты соответствуют основным консервативным генам морбилливирусов, сохранившим коллинеарное расположение. Разрывы между сегментами, вероятно, представляют собой вариабельные межгенные участки или гипервариабельные домены внутри генов.
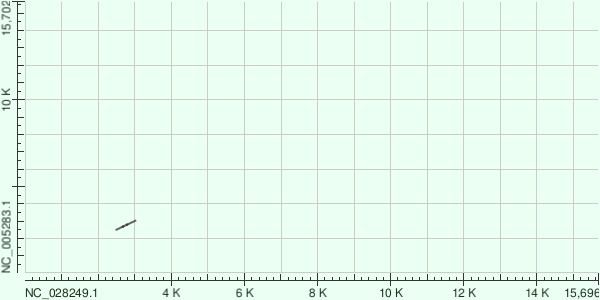
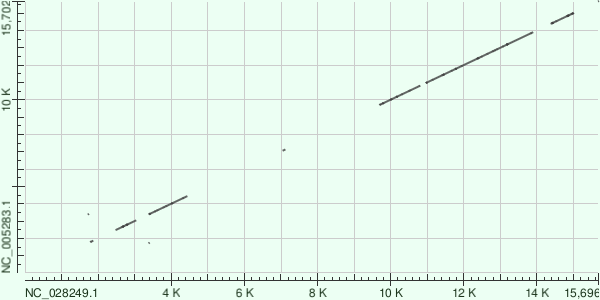
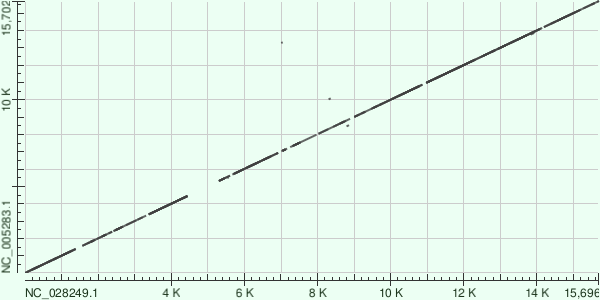
Карта BLASTN (стандартные параметры)
Карта, построенная алгоритмом BLASTN, демонстрирует наиболее полную и плотную прерывистую диагональ среди всех нуклеотидных сравнений. Алгоритм blastn, оптимизированный для поиска дивергентных последовательностей, оказался чувствительнее, чем megablast с word size=16. Он выявил больше коротких гомологичных участков, которые были пропущены Megablast.
Карта TBLASTX (стандартные параметры)
Карта от TBLASTX кардинально отличается: она показывает практически непрерывную и чёткую главную диагональ. Поскольку TBLASTX сравнивает шесть потенциальных рамок считывания на уровне аминокислотных последовательностей, этот результат доказывает, что, несмотря на нуклеотидные различия, белковые продукты данных геномов высококонсервативны. Сплошная диагональ также подтверждает отсутствие крупных геномных перестроек (инверсий, транслокаций)