Практикум 12
Множественное выравнивание последовательностей белков
Автор старался, но не может гарантировать отсутствие биологических ошибок.
2. Сравнение выравнивания одних и тех же последовательностей разными программами
Было решено продолжить работу с представителями семейства доменов RNR_Alpha - им посвящался практикум 11. В UniProt были найдены 14 белков, содержащиеся в базе данных Swiss-Prot и в последовательности которых обнаружен домен PF17975: (xref:pfam-PF17975) AND (reviewed:true). Скачены их fasta последовательности: файл. Запустим Jalview. Проведем выравнивание различными предлагаемыми в Web Allignment программами. Получившиеся fasta-файлы с выравниванием: Tcoffee, Probcons, Muscle, Mafft, MSAprobs, GLprobs, Clustal, ClustalO. Сравним программы множественного выравнивания, используя сервис VerAlign. Метод подсчета очков был выбран совместный: подсчет очков за колонки, подсчет очков за совпадание пар номеров букв в последовательности (самих букв естественно тоже). Остальные параметры оставлены по умолчанию. VerAlign был запущен для каждой пары программ MSA, очки записаны в таблицу excel. Затем был проведен анализ полученных результатов - рисунки 1, 2.
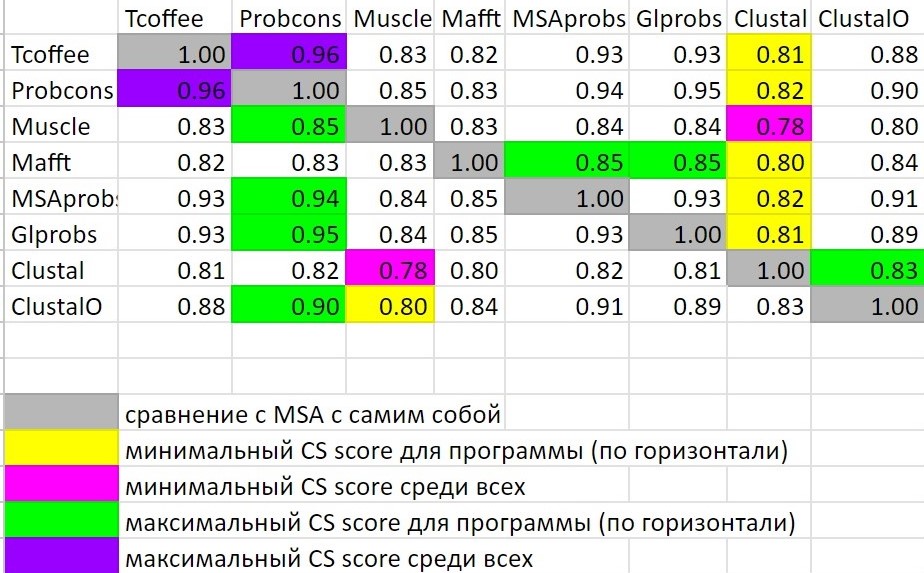
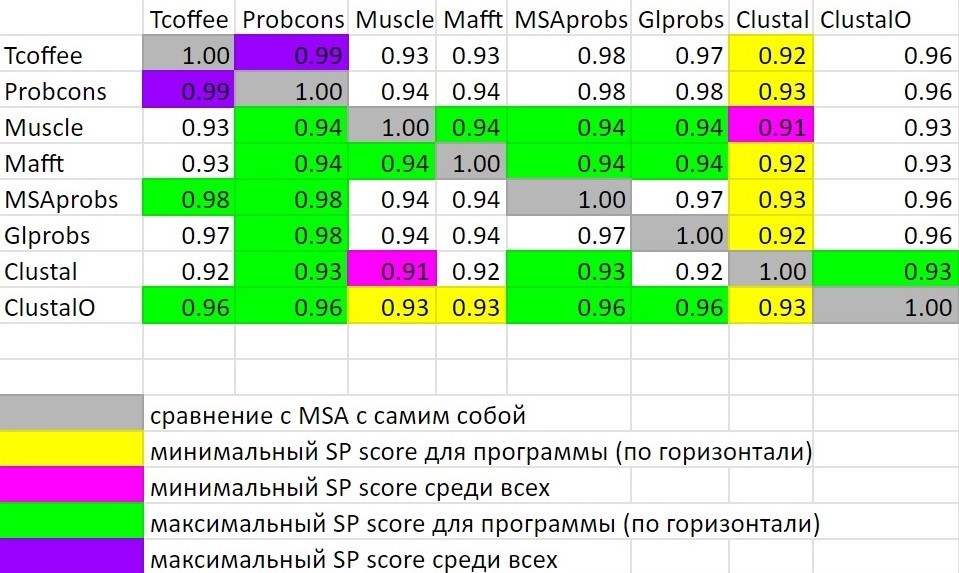
Видно, что критерий совпадающих колонок является более строгим чем критерий совпадающих пар номеров букв в последовательностях. В данном случае, результаты Clustal больше всего отличаются от результатов остальных программ множественного выравнивания; выравнивание Probcons, наоборот, представляет собой компромисс множественных выравниваний. Было проведено более детальное сравнение Clustal и Muscle (наиболее отличающиес) - проект Jalview (рис 3), Tcoffee и Probcons (самые схожие) - проект Jalview (рис 4). Уже "глазками" видно, что столбцы выравнивания совпадают во втором случае намного чаще и одинаковые столбцы находятся ближе друг к другу. Пример для результатов программ Clustal и Muscle совпадающего участка: 96-110, несовпадающих колонок: 135 -156. Аналогично для результатов программ Tcoffee и Probcons: 114-224 и 225-243. Даже при случайно подобранных участках заметно, что для первой пары программ множественного выравнивания совпадения в выдаче происходят реже.
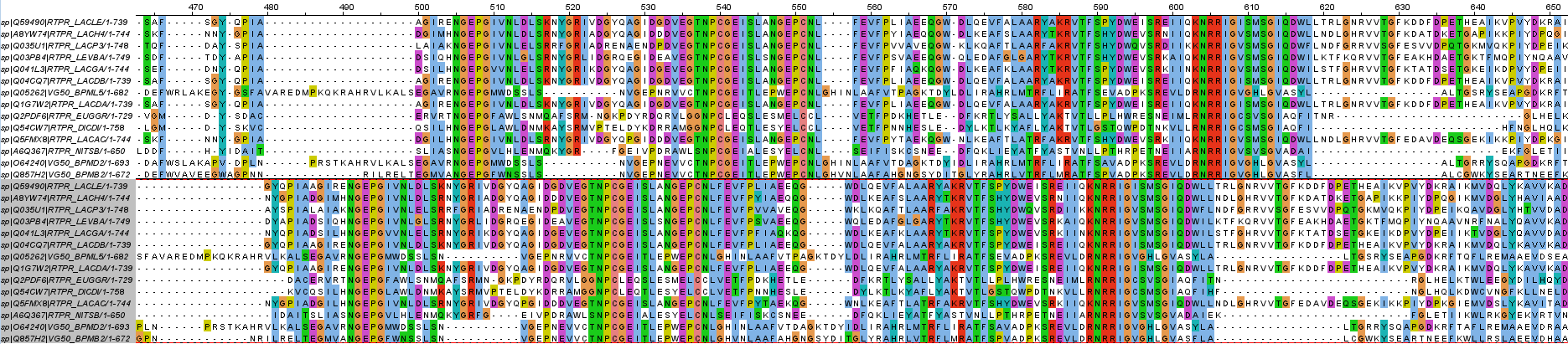

3. Построение выравнивания по совмещению структур и сравнение его с выраваниванием MSA
Для выполнения задания было выбрано семейство ABC-транспортеров (ABC transporter) -
ABC_tran (PF00005). Семейство принадлежит семейству АТФ-связывающих
кассет (ATP-Binding Cassette superfamily). Обычно молекулы состоят из имнимум двух регионов: АТФ-связывающей кассеты и
трансмембранного домена (TMD). Второй, как правило, менее консервативен.
Представители семейства (таблица 1) были найдены в базе UniProt (выбраны проверенные белки с известной 3D-структурой):
(xref:pfam-PF00005) AND (reviewed:true) AND (existence:1).
Для выравнивания использовался онлайн-инструмент pairwise structure alignment на RCSB PDB. Выравнивание 3D-структур в PDB:
ссылка.
Далее были скачены 3D-структуры в формате .cif и открыты в PyMol, результаты сравнений 3D-структур представлены на рисунках 5-8.
| AC | ID | Organism | PDB |
|---|---|---|---|
| Q9NRK6 | ABCBA_HUMAN | Homo sapiens (Human) | 3ZDQ |
| P77499 | SUFC_ECOLI | Escherichia coli (strain K12) | 2D3W |
| P40024 | ARB1_YEAST | Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) | 6R84 |
| Q9JJ59 | ABCB9_MOUSE | Mus musculus (Mouse) | 7V5C |
| ABCB25 | AB25B_ARATH | Arabidopsis thaliana (Mouse-ear cress) | 7N58 |
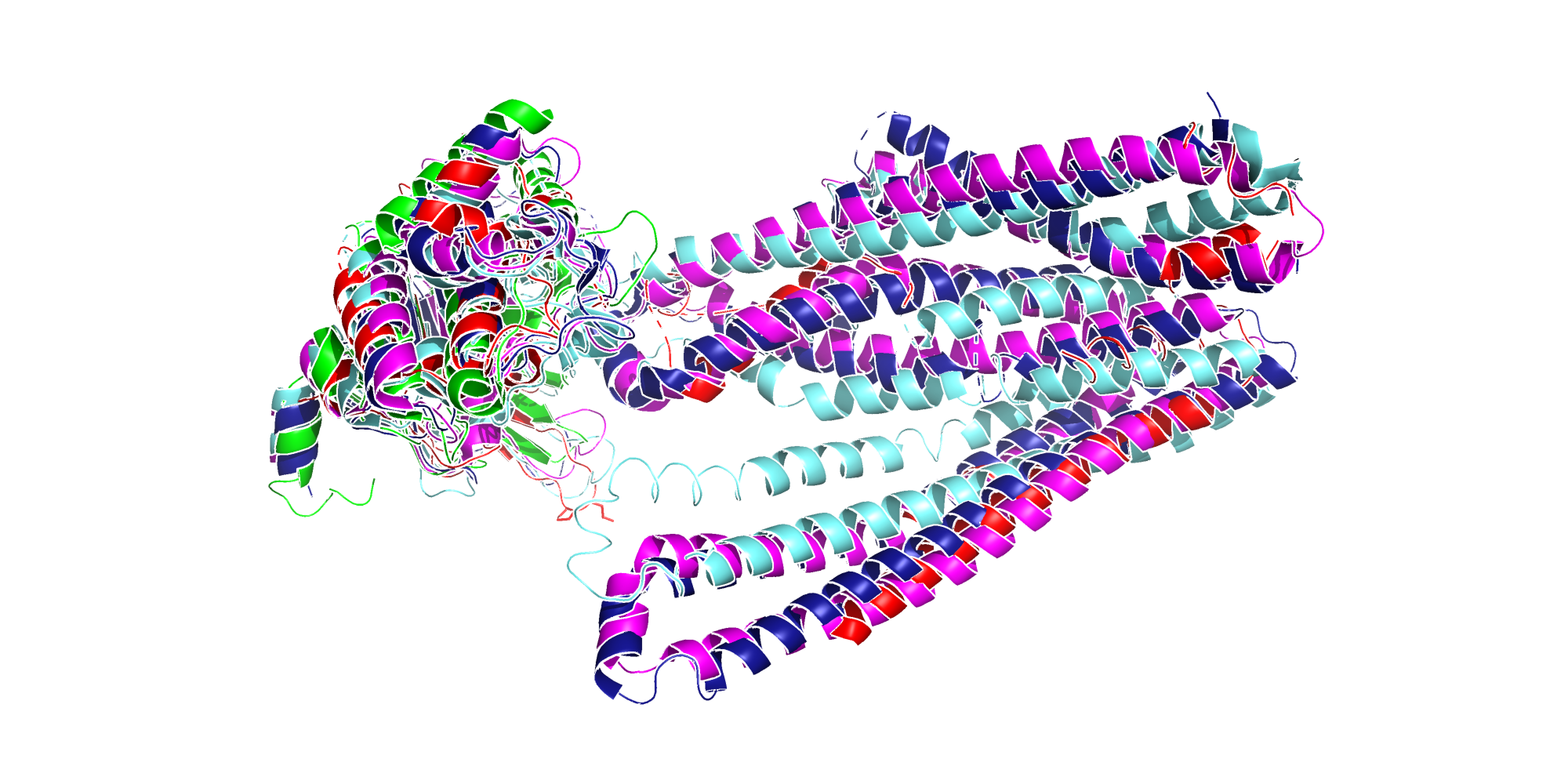
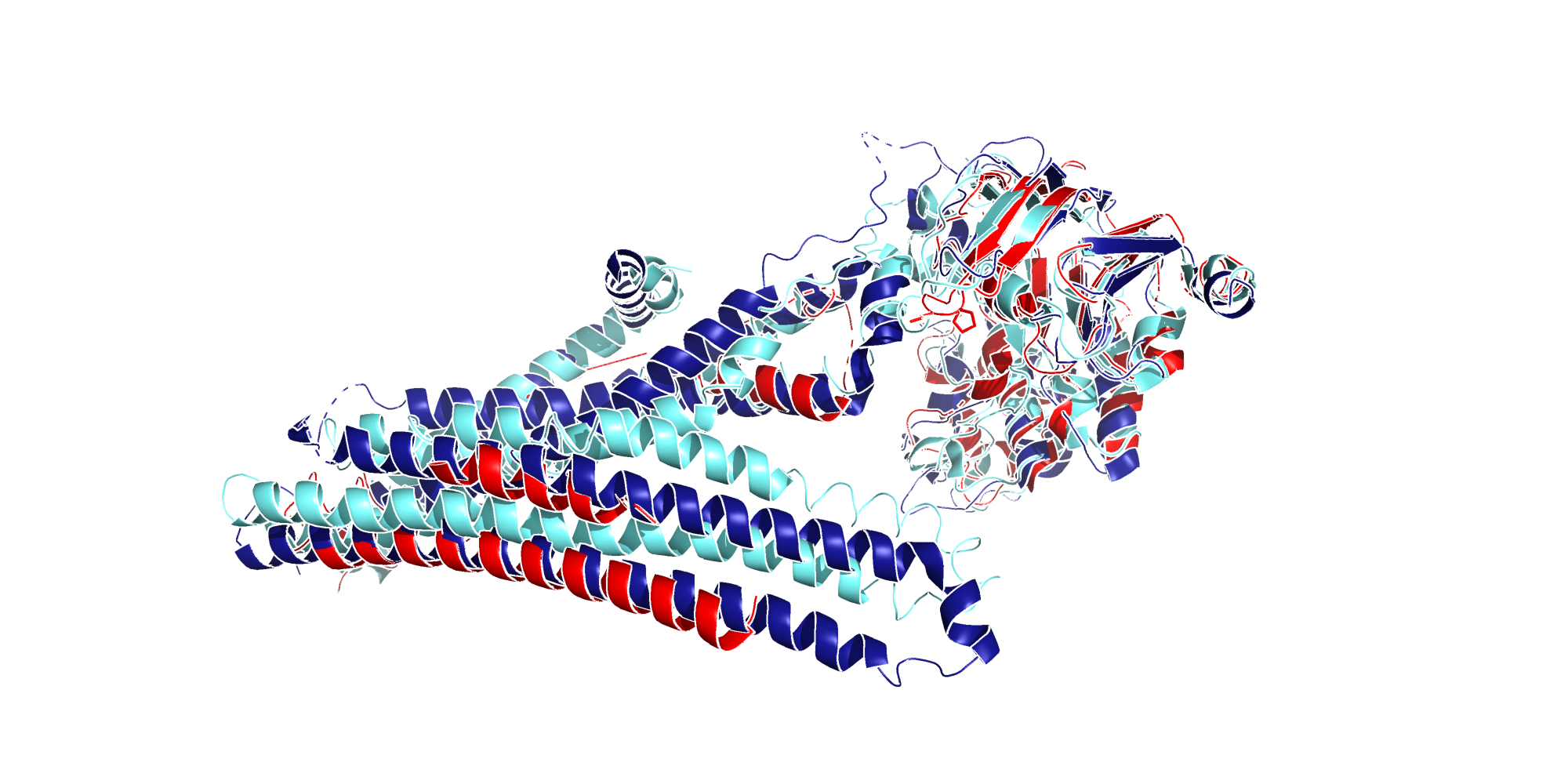
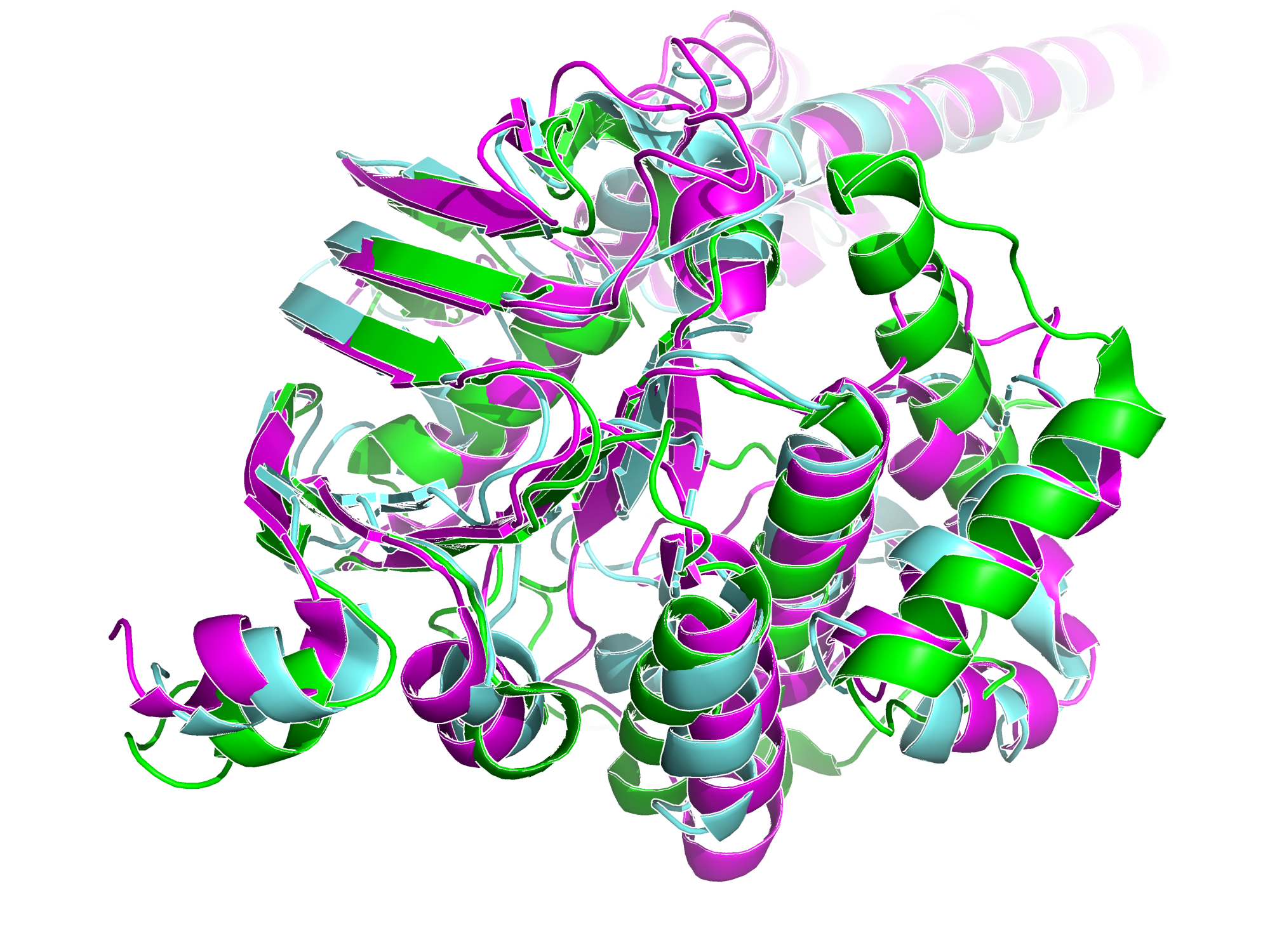
Заметно, что 3D-структура белка для бактерии (E.coli) меньше, что скорее связано с тем, что домены ABC-транспортеров иногда разделяются по разным цепям, а для анализа всех организмов отбирались только A цепи соответствующих молекул (рис 5). По современной систематике принято относить растений к надцарсту Archaeplastida, грибы и животные же выделяются в Amorphea, поэтому удивительно, что структуры выбранных белков более схожи для растения и дрожжей, чем для мышки и дрожжей (рис 6). Более ожидаемым было сходство структур для мышки и человека (рис 7), а также дрожжей и растений (рис 8), по сравнению с пространственной конфигурацией для бактериального белка, ведь все выбранные организмы относятся к эукаритам, кроме E.coli, являющейся представителем прокариот.
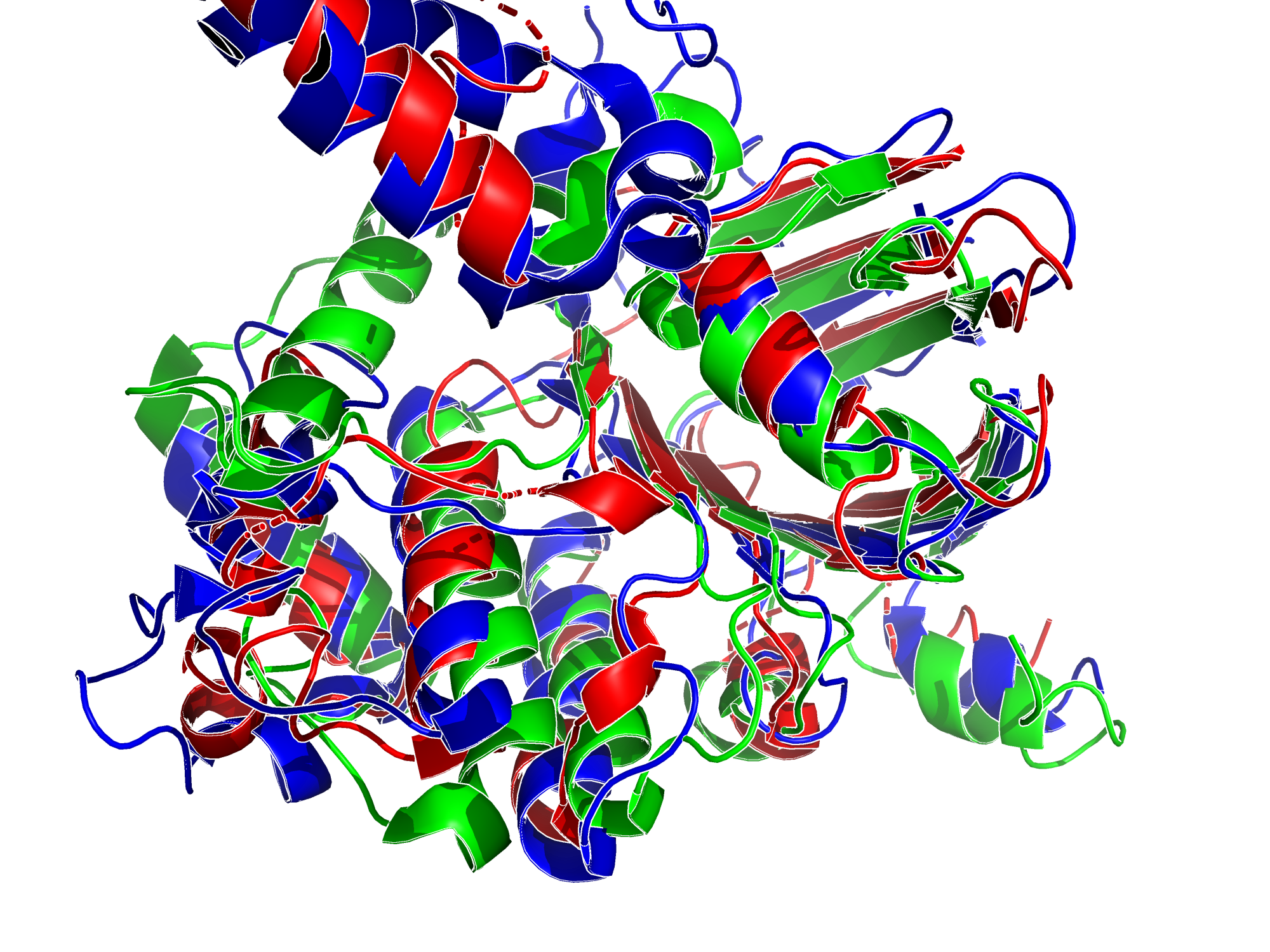
Из сервиса выравнивания на RCSB PDB также был скачен fasta-файл выравнивания. К сожалению, там была представлена совокупность парных выравниваний с первым белком (в данном случае - белок человека, 3ZDQ). Проект Jalview первоначальный, где можно наблюдать эти парные выравнивания. Ручное подравнивание четырех последовательностей предполагает большую вероятность возникновения ошибки из-за человеческого фактора, поэтому для получения удобного вида множественного выравнивания написан скрипт на Python. Был получен новый fasta-файл, содержащий выравнивание 5 последовательностей по совмещению пространственных структур. Проекты Jalview с последовательностями целиком, с выделенным основным доменом (АТФ-связывающая кассета). Затем были удалены все гэпы и последовательности были выравнены программой Muscle с настройками по умолчанию. Jalview проекты с результатом для всей последовательности, с выделенным фрагментом. Выравнивания можно сравнить "глазками" на рисунках 9,10, а также в проекте Jalview. Основной тренд - в выравнивании на основе пространственных структур индели значительно длиннее и чаще, чем при выравнивании программой Muscle. Сервис Veralign выдавал ошибку при попытке сравнить два выравнивания, возможно, потому что они отличаются по длине на приблизительно 160 колонок.


4. Краткое описание Muscle - программы MSA
Общая информация
MUSCLE (Multiple Sequence Comparison by Log-Expectation) - программа
для множественного выравнивания последовательностей аминокислот и нуклеотидов.
Первые статьи про программу появились еще в 2004 году.
Считается, что при правильном подборе опций Muscle способна получать результаты быстрее и точнее,
чем ClustalW и TCoffee. В целом даже при опциях по умолчанию,
MuscleV5 генерирует высокоточное выравнивание. Программа установлена и на kodomo (пример использования -
практикум 9), принимает на вход различные опции, из которых обязательная -in
'входной файл fasta-формата', помощь можно вызвать по man muscle или muscle -help.
Алгоритм
Выделяют три стадии выравнивания в MUSCLE: черновик прогрессивного выравнивания, улучшенное прогрессивное выравнивание и
стадия отладки.
Стадия 1. Строится бинарное дерево на основе матрицы расстояний. Для каждого узла в дереве, начиная с
"листьев", строится парное выравнивание. По достижении "ствола" дерева имеется множественное выравнивание всех
входных последовательностей. На этой стадии основной упор на скорость, а не на точность.
Стадия 2. На основе имеющегося дерева и множественного выравнивания строится новая матрица расстояний.
Затем как и на 1 стадии строится множественное выравнивание, при этом выравнивания для неизменных пар
не пересчитываются. Получаем более оптимальное дерево и увеличивается точность множественного выравнивания.
Стадия 3. Рекурсивно выполняется следующий порядок действий до достижения конвергентности или лимита,
установленного пользователем:
1) выбирается "ветка" дерева из второй стадии
2) выбранная "ветка" выделяется в качестве самостоятельного дерева и удаляется из первоначального дерева -
получаем два под-дерева;
3) профиль множественного выравнивания вычисляется для каждого из под-деревьев;
4) перевыравниваются выравнивания из каждого под-дерева - получаем новое множественное выравнивание;
5) происходит оценка нового множественного выравнивания, если у него больше пар совпадающих позиций (SP score),
то новое выравнивание сохраняется, иначе - удаляется.
"Ветки" отбираются в порядке увеличения расстояния от "ствола".
Список литературы
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004 Mar 19;32(5):1792-7. doi: 10.1093/nar/gkh340. PMID: 15034147; PMCID: PMC390337.
- Edgar, R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004). https://doi.org/10.1186/1471-2105-5-113
- Official page
- Online EMBL version
- Основной источник информации