Поиск полиморфизмов у человека
Подготовка чтений
Работаем с хромосомой 19.
Сделали контроль качества чтений с помощью программы FastQC. На kodomo вызываем программу командой fastqc сhr19.fastq
Программа обрабатывает чтения из файла chr19.fastq и выдает архив с отчетом о работе в формате html.
Отчет FastQC
С помощью программы Trimmomatic провели очестку чтений.
Удалили с конца каждого чтения нуклеотиды с качеством ниже 20 TRAILING:20;
оставили только чтения длиной не меньше 50 нуклеотидов MINLEN:50.
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr19.fastq chr19_out.fastq TRAILING:20 MINLEN:50
Программа прочитала 5524 рида и удалила из них 297, видимо, те у которых длина меньше 50 нуклеотидов.
Проанализировали программой FastQC качество чтений после чистки.
Отчет FastQC
Посмотрим раздел "Per base quality" в выдаче FastQC до и после чистки.
| Per base quality |
| До чистки |
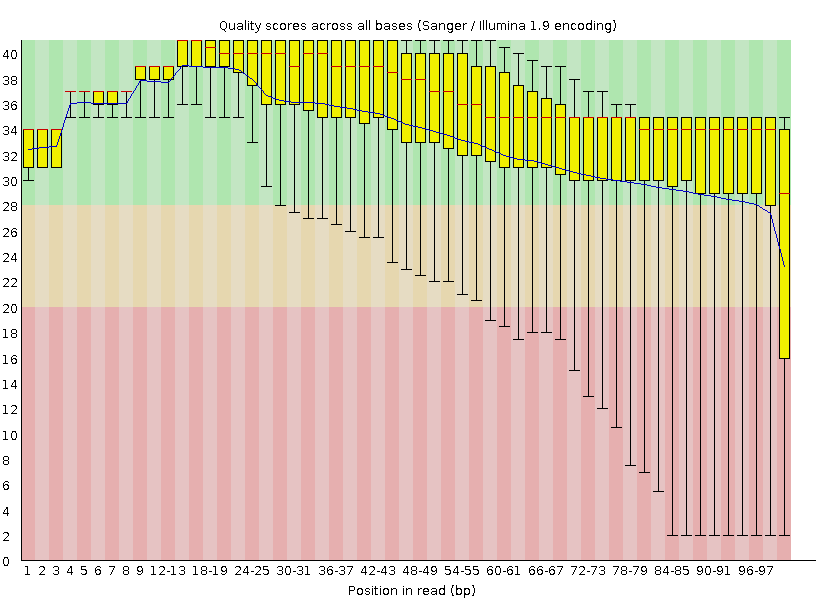 |
| После чистки |
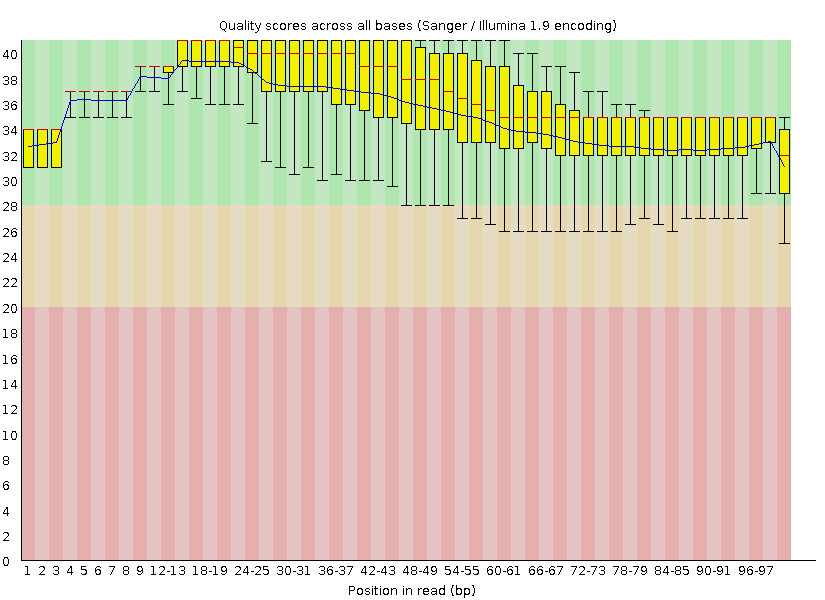 |
|
Даная картинка отражает изменение качества чтений до и после очистки с помощью программы Trimmomatic По оси Х отложены позиции в риде, по оси У – оценки качества прочтения нуклеотидов Quality scores. Для каждой позиции посроена диаграмма размахов (BoxWhisker type plot). Центральная красная линия обозначает медиану, желтый прямоугольник - интерквартильный рахмах (25-75%), концы усов — края статистически значимой выборки (в данном случае - точки 10% и 90%), синяя линия - математическое ожидание (mean) по качеству. График разделен на три области: зеленую – высокая точность прочтения нуклеотида, оранжевую – приемлимая точность, красную – низкая точность. Как видно на первой картинке точность прочтения нуклеотида постепенно снижается. Последние нуклеотиды читаются с более низкой точностью. Качество прочтения некоторых попадает в красную зону. После очистки с концов чтений были удалены нуклеотиды с качеством ниже 20. Как мы видим размах усов уменьшился. Качество прочтения концевых нуклеотидов не опускается ниже 24. Кроме того, синяя линия на втором графике расположена выше, т.е. повысилось среднее арифметическое качества прочтения нуклеотидов. Таким образом, в результате очистки чтений повысилось их качество. |
Картирование чтений и анализ выравнивания
С помощью программы BWA откартировали очищенные чтения.
- Проиндексировали референсную последовательность
- bwa index chr19.fasta
- Построили выравнивание прочтений и референса в формате .sam, используя алгоритм mem
- bwa mem chr19.fasta chr19_out.fastq > align.sam
- С помощью пакета samtools перевели выравнивание чтений с референсом в бинарный формат .bam.
- samtools view align.sam -b -o align.bam
- Отсортировали выравнивание чтений с референсом по координате в референсе начала чтения
- samtools sort align.bam -o align_sort.bam -T tmp.txt
Опция -T, чтобы во время работы программы данные записывались во временный файл, а не выводились в stdout. - Проиндексировали отсортированный .bam файл
- samtools index align_sort.bam
- Выяснили, сколько чтений откартировалось на геном
- samtools idxstats align_sort.bam > align_stat.out
Содержимое файла align_stat.out:
- Название последовательности
- chr19
- Длина последовательности
- 59128983
- Число картированных ридов
- 5227
- Число некартированных ридов
- 0
Анализ SNP
- Создали файл с полиморфизмами в формате .bcf
- samtools mpileup -uf chr19.fasta align_sort.bam -o snp.bcf
- Создали файл со списком отличий между референсом и чтениями в формате .vcf.
- bcftools call -cv snp.bcf -o snp.vcf
Файл snp.vcf содержит список отличий между референсом и ридами в формате .vcf. Найдено 92 полиморфизма: 82 замены и 6 инделей. Четыре полиморфизма описаны в таблице 2.
Таблица 2. Описание четырех найденных полиморфизмов
| Координата (POS) |
Тип полиморфизма | Последовательность референса (REF) |
Последовательность в ридах (ALT) |
Качество чтений (QUAL) |
Глубина покрытия (DP) |
|---|---|---|---|---|---|
| 17302112 | делеция | ACTCTCT | ACTCT | 69.4665 | 6 |
| 45395619 | замена | A | G | 225.009 | 124 |
| 45406538 | вставка | CGGGGGGG | CGGGGGGGG | 25.4748 | 22 |
| 18304700 | замена | A | G | 222 | 36 |
Мы проанализировали покрытие и качество всех найденных полиморфизмов. Данные, полученные с помощью Excel, представлены в таблице 3.
У некоторых участков хорошее покрытие и, следовательно, хорошее качество. Некоторые участки встречаются в чтениях только по 1-2 раза, качество этих участков низкое.
Таблица с рассчетами
Таблица 3. Данные о качестве находок
| Quality | Depth | |
|---|---|---|
| min | 3,0136 | 1 |
| max | 225,009 | 68 |
| average | 84,487 | 11,71739 |
| median | 64,5073 | 8 |
Аннотация полиморфизмов
С помощью программы annovar проаннотировали полученные snp по базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
С помощью скрипта convert2annovar.pl. мы получили из файла snp.vcf получили файл, с которым умеет работать программа annovar.
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput
Для аннотации файла с snp с помощью предложенных баз данных использовали скрипт annotate_variation.pl.
Аннотация по базе refgene – gene-based annotation
perl /nfs/srv/databases/annovar/annotate_variation.pl -out snp.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
Программа вернула три файла:
- snp.refgene.variant_function (содержит описание всех SNP);
- snp.refgene.exonic_variant_function (содержит описание SNP, попавших в экзоны);
- snp.refgene.log (содержит информацию о процессе работы).
В таблице 4 перечислены катагории, на которые разделяются snp в базе refseq.
Таблица 4. Категории snp [1]
| Значение | Расшифровка |
|---|---|
| exonic | Полиморфизм внутри экзона |
| splicing | полиморфизм в пределах 2 bp от границы сплайсинга |
| ncRNA | полиморфизм в последовательности, не аннотированной как кодирующая. |
| UTR5 | полиморфизм в 5' нетранслируемом регионе |
| UTR3 | полиморфизм в 3' нетранслируемом регионе |
| intronic | полиморфизм в интроне |
| upstream | полиморфизм в пределах 1-kb upstream от сайта начала транскрипции |
| downstream | полиморфизм в пределах 1-kb downstream от сайта окончания транскрипции (можно изменить этот параметр опцией -neargene) |
| intergenic | полиморфизм на пересечении генов |
Изучив файл snp.refgene.variant_function, мы можем сказать, что в данных ридах всего 86 snp. Из них: 4 UTR3; 12 exonic; 70 intronic; 35 гомозиготных замен 51 гетерозиготных.
Некодирующие последовательности могут накапливать мутации, т.к. эти последовательности не подвегаются отбору. Замены в кодирующей последовательности могут привести к несинонимичной замене в аминокислотной поледовательности и, как следствие, к нарушению структуры и функции белка. Поэтому exonic snp встречаются гораздо реже, чем intronic snp.
В файле snp.refgene.exonic_variant_function представлена информация о полиморфизмах в экзонах. Всего 12 таких замен. Замены могут быть синонимичными, т.е. замена нуклеотида не приводит к изменению аминокислоты. Это возможно, т.к. генетический код вырожден – некоторым аминокислотам соответствует несколько кодонов. Несинонимичные замены, приводящие к замене аминокислоты, могут приводить к изменениям структуры и фукци белка. Из 12 замен 3 несинонимичных.
Таблица 5. Некоторые замены
| № | Координаты | Ген | Нуклеотидная замена | Тип замены | Синонимичность | Качество чтений | Глубина покрытия | АК замена |
|---|---|---|---|---|---|---|---|---|
| 1 | 17303774 | MYO9B | T>G | exonic, het | nonsynonymous | 103.008 | 10 | S>A |
| 2 | 17316782 | MYO9B | T>C | exonic, het | nonsynonymous | 73.0074 | 10 | V>A |
| 3 | 18304700 | MPV17L2 | A>G | exonic, hom | nonsynonymous | 222 | 36 | M>V |
| 4 | 45395714 | TOMM40 | T>C | exonic, het | synonymous | 225.009 | 68 | F>F |
Замены произошли в генах следующих белков:
MYO9B – Миозин IXB (произошло 2 несинонимичных замены в кодирующей последовательности)
MPV17L2 (1 несинониминая замена)
TOMM40 транслоказа наружней митохондриальной мембраны.
Аннотация по базе dbsnp – filter-based annotation
perl /nfs/srv/databases/annovar/annotate_variation.pl -out snp.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Узнать, сколько полиморфизмов имеют rs мы можем из файлов snp.dbsnp.hg19_snp138_droppedsnp.dbsnp.hg19_snp138_filtered. В файл "filtered" попало 10 snp, не имеющих rs, т.е. неаннотированных в базе dbsnp. Все "отфильтрованные" полиморфизмы имеют покрытие 3 и менее и низкое качество прочтения. B файл "dropped" попало 76 snp, у которых есть rs, значит, они аннотированы в данной базе. Качество и глубина покрытия этих snp разнообразны (от минимального до максимального)
Аннотация по базе 1000 genomes – filter-based annotation
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Программа выдала несколько файлов. В файл snp.1000.hg19_ALL.sites.2014_10_filtered попало 11 snp, не аннотированных в базе 1000 genomes. Остальные 75 попали в файл snp.1000.hg19_ALL.sites.2014_10_dropped. Для них можем узнать частоту (во второй колонке). Частота аннотированных полиморфизмов варьирует от 0,0002 до 0,780751, средняя частота 0,4185
Средняя частота для экзонных snp выше (0,470). [Данные получены с помощью рассчетов в Excel таблица ]Аннотация по базе Gwas – region-based annotation
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out snp.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
В файле snp.gwas.hg19_gwasCatalog перечислены snp, аннотированные в базе Gwas. Мы можем узнать, какие из найденных полиморфизмов имеют клиническое значение. У нашего пациента найдено 4 snp, имеющих клиническое значение. Данные о них представлены в таблице 6.
Таблица 6. Описание клинически значимых полиморфизмов пациента по данным базы Gwas
| № | Координата | Ген | Категория | Клиническое значение |
|---|---|---|---|---|
| 1 | 17283303 | MYO9B | intronic | Связан с ростом |
| 2 | 18304700 | MPV17L2 | exonic | Рассеянный склероз[2] |
| 4 | 45395619 | TOMM40 | intronic | Биомаркер болезни Альцгеймера[3] |
| 3 | 45396219 | TOMM40 | intronic | Метаболический синдром[4] |
Среди этих замен только одна экзонная. Мутации в интронах также могут иметь клиническое значение. Например они могут влиять на сплайсинг белка и таким образом вызывать нарушения в последовательности белка.
Аннотация по базе Clinvar filter-based annotation.
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out snp.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
Все 86 snp оказались в файле snp.clinvar.hg19_clinvar_20150629_filtered, т.е. такие полиморфизмы не аннотированы в базе Clinvar.
Сводная таблица
В сводной таблице собрана информация обо всех snp, которую мы получили из разных баз данных в ходе практикума. Красным выделены строки с полиморфизмами, имеющими клиническое значение по данным базы Gwas. Т.к. ни одна из данных 86 замен не аннотирована в Clinvar, соответствующей колонки в таблице нет.