Ресеквенирование. Поиск полиморфизмов у человека.
Для работы мне были предоставлены файлы
chr11.fastq и chr11.fasta.
Все действия выполнялись в
специально созданной директории /nfs/srv/databases/ngs/ann_karpukhina1
Анализ качества чтений
Программа FastQC позволяет проводить быстрый и несложный контроль качества "сырых" данных секвенирования. Она проводит
набор стандартных статистических анализов, которые можно использовать для предварительной оценки релевантности
данных и выявления возможных проблем в полученной последовательности.
Для проведения анализа качества чтений была иcпользована FastQC,
установленная на Kodomo. Для удобства
работы я также установила на свой компьютер версию с графическим интерфейсом.
Использованная команда: fastqc chr11.fastq
В результате был получен архив chr11_fastqc.zip, содержащий отчет о работе программы в виде html файла -
chr11_fastqc.html.
В целом качество чтений можно назвать уловлетворительным, однако для повышения качества лучше провести чистку.
Очистка чтений
Очистка чтений была проведена с помощью программы
Trimmomatic. Адаптеры в исследуемой последовательности
были уже удалены. Необходимо было отсечь с конца чтения нуклеотиды с неудовлетворительным качеством (< 20), для этого был указан параметр
TRAILING:20. Также требовалось оставить только прочтения длины не менее 50, поэтому был указан
параметр MINLEN:50.
Использованная команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11.fastq chr11_trim.fastq TRAILING:20 MINLEN:50
Выходной файл: chr11_trim.fastq
Сравнение качества до и после чистки
"Очищенная" последовательность была проанализирована с помощью
FastQC.
Использованная команда: fastqc chr11_trim.fastq
Выходной файл: chr11_trim_fastqc.html
Далее было проведено сравнение качества исходной и очищенной последовательности, представленное в таблице ниже.
Слева - данные по исходному файлу, справа - по отредактированному Trimmomatic.
Галочка, восклицательный знак или крестик рядом с рисунком обозначают отсутсвие или наличие соответсвующего предупреждения
FastQC.
| Сравнение качества до и после чистки | |
|---|---|
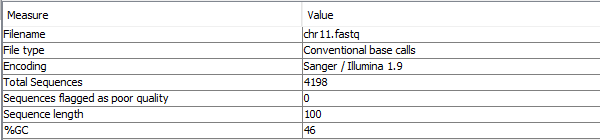 | 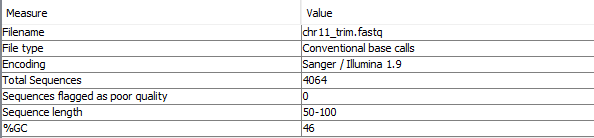 |
| Рис. 1. ✔ Basic statistics, chr11.fastq | Рис. 2. ✔ Basic statistics, chr11_trim.fastq |
| После очистки число ридов уменьшилось на 134 (с 4198 до 4064), были удалены риды наиболее низкого
качества. Остальные параметры остались без изменений. |
|
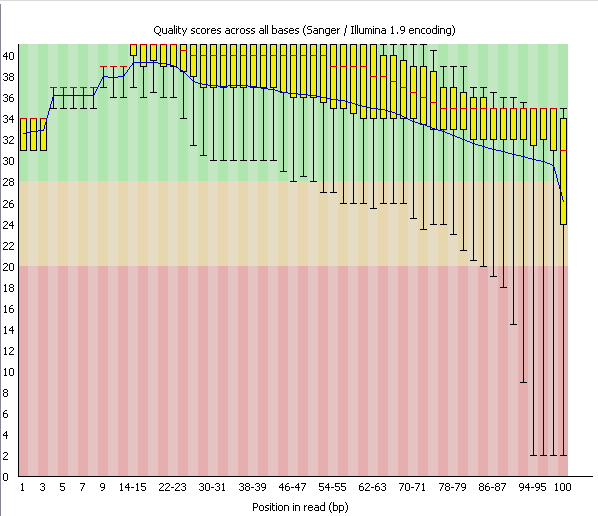 | 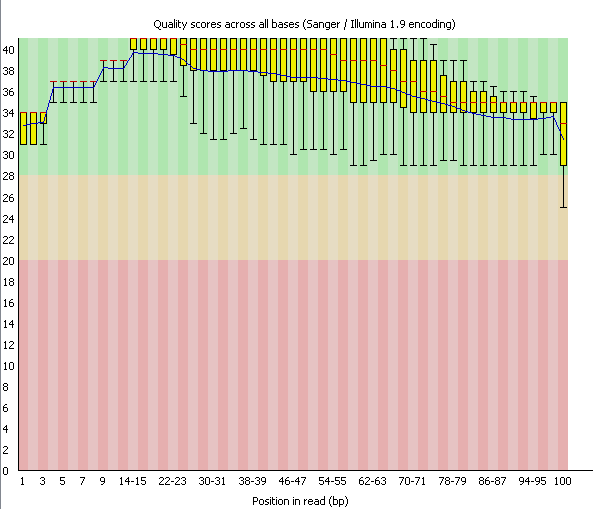 |
| Рис. 3. ✔ Per Base Sequence Quality, chr11.fastq | Рис. 4. ✔ Per Base Sequence Quality, chr11_trim.fastq |
| Per Base Sequence Quality показывает разброс качества по всем основаниям в каждой позиции
FastQ файла. Для каждой позиции построена диаграмма размахов (BoxWhisker type plot, ящик с усами). Центральная красная линия обозначает
медиану, желтый прямоугольник - интерквартильный рахмах (25-75%), концы усов — края статистически значимой выборки (в данном случае -
точки 10% и 90%), синяя линия - математическое ожидание (mean) по качеству. На оси OY показаны оценки качества (quality scores). Чем выше quality score, тем точнее определено основание. Фон графика разделен на три области: очень хорошая точность (зеленый цвет), приемлемая точность (оранжевый), низкая точность (красный). Для большинства платформ качество постепенно понижается со временем от начала секвенирования, поэтому достаточно типичным является попадание прямоугольников/усов в оранжевую область ближе к концу рида. Это мы можем наблюдать и в нашей последовательности, особенно в той, что не подвергалась очистке. В случае, когда хотя бы для какого-то из оснований нижний квартиль меньше 10 или медиана меньше 25, программа выдает предупреждение о том, что с последовательностью могут быть проблемы (warning). Если нижний квартиль меньше 5 или медиана меньше 20, программа выдает сообщение об ошибке в последовательности (failure). Для нашей последовательности Per Base Sequence Quality было приемлемым как до, так и после чистки (программа не выдавала предупреждение). Однако можно заметить, что после чистки значительно уменьшилось число усов, выходящих из зеленой области (до чистки они составляли примерно половину, а после остался всего один). Медиана и в том, и в другом случае осталась примерно такой же, а вот математическое ожидание после чистки повысилось. Все это говорит о более высоком качестве оставшихся чтений. Таким образом, Trimmomatic вырезал низкокачественные чтения и оставил только те, с которыми можно нормально работать. |
|
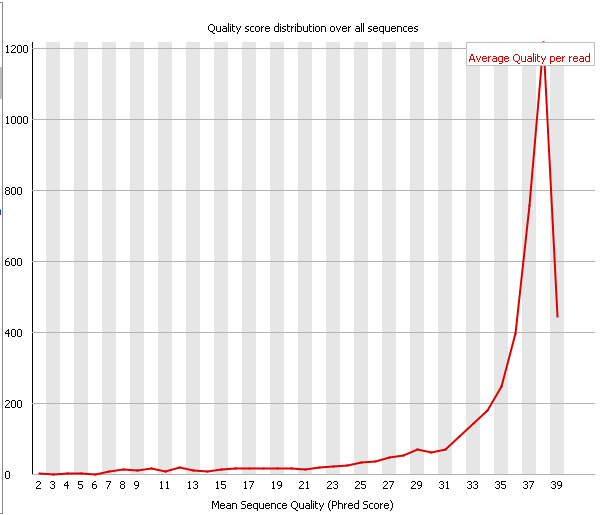 | 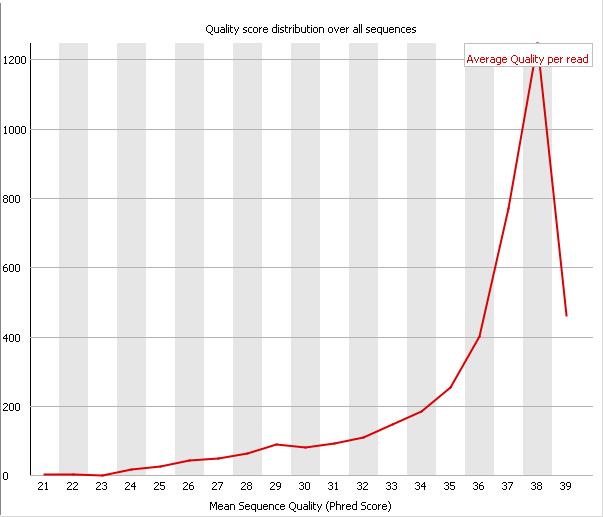 |
| Рис. 5. ✔ Per Sequence Quality Score, chr11.fastq | Рис. 6. ✔ Per Sequence Quality Score, chr11_trim.fastq |
| Per Sequence Quality Score позволяет определить наличие подмножеств ридов с выделяющимся общим низким качеством.
Целое подмножество ридов обычно имеет низкое качество из-за плохого качества соответсвующего изображения (например на краю
видимой области).Тем не менее такие риды, как правило, представляют собой лищь небольшой процент от общего числа, поэтому
серьезные отклонения в этом модуле встречаются относительно редко. Warning выдается, когда среднее значение качества
опускается ниже 27, что соответсвует 0.2% вероятности ошибки. В случае, когда среднее ниже 20 (вероятность ошибки 1%), программа
выдает Failure. Как до, так и после чистки Per Sequence Quality Score для нашей последовательности был стабильным. После чистки число ридов уменьшилось, поэтому уменьшилось и число делений на оси OX (была обрезана область самого низкого качества).Однако среднее значение качества на рид осталось прежним - 38. Это достаточно хороший показатель. |
|
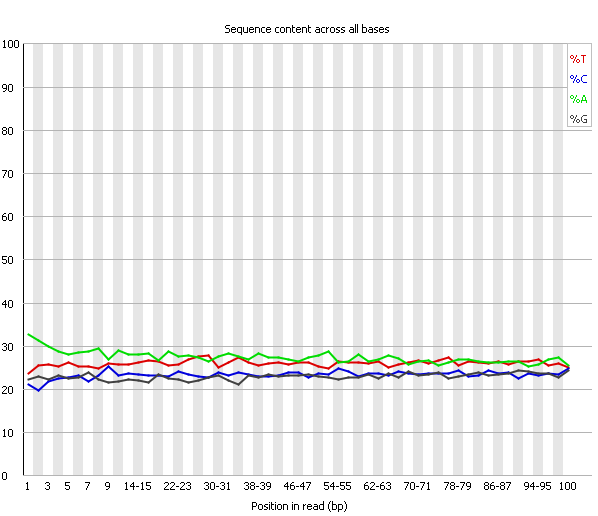 | 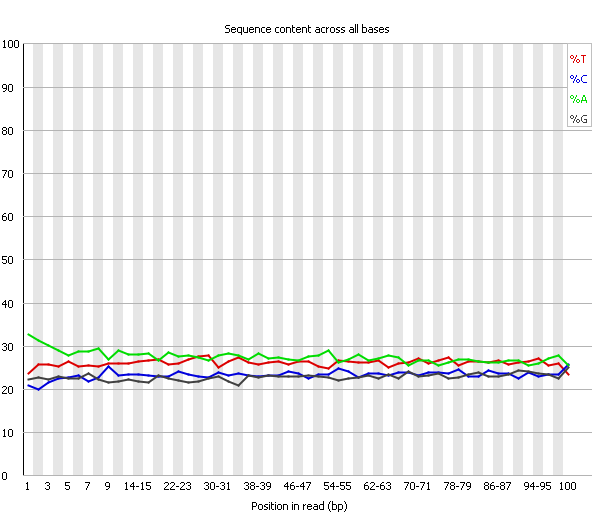 |
| Рис. 7. ❗ Per Base Sequence Content, chr11.fastq | Рис. 8. ❗ Per Base Sequence Content, chr11_trim.fastq |
| Per Base Sequence Content графически отображает относительные частоты появления каждого из 4-х оснований в
каждой позиции. В случайной библиотеке ожидается, что различия во встречаемости различных оснований от цикла к циклу будут
крайне малы или вовсе равны нулю, соответсвенно линии на графике должны быть параллельны. Относительное количество каждого
из оснований должно отражать общее количество данных оснований в геноме, поэтому строгая параллельность обычно не наблюдается,
но в любом случае отклоения прямых друг от друга не должны быть очень велики. В случае, когда в какой-либо из позиций
различия между встречаемостью A и T или G и С превышают 10%, выдается Warning, 20% - Failure. Для нашей последовательности как до, так и после чистки, программа выдавала предупреждение. Вероятнее всего причиной послужило большое различие между A и Т в начальных позициях (см. Рис. 7-8). Тем не менее, в Help для FastQC отмечено, что в триммированных по адаптерам последовательностях (а у нас были именно такие последовательности) бывает так, что сиквенсы, у которых на концах случайно оказалась часть, комплементарная части адаптера, ошибочно обрезаются. Таким образом, остаются только сиквенсы, концы которых не комплементарны адаптерам, что и может вызвать мнимый диссонанс в относительных количествах оснований. Возможно, у нас именно такой случай, хотя точно ничего утверждать нельзя. Существенных различий между показателями до и после чистки не заметно. |
|
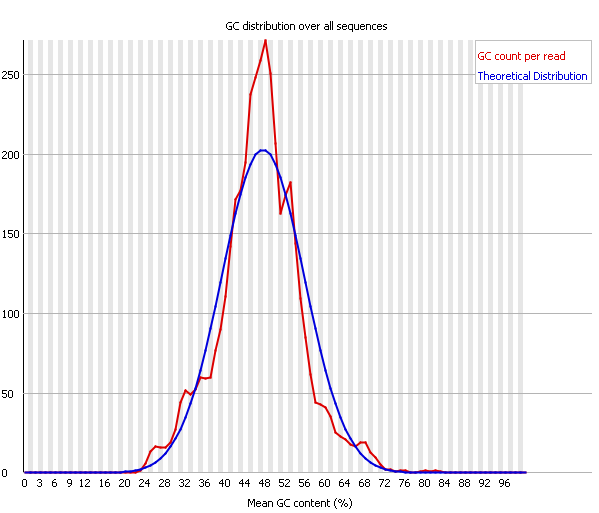 | 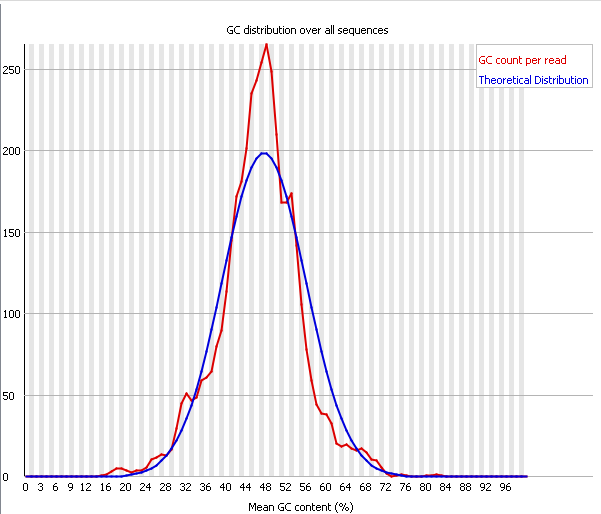 |
| Рис. 9. ❗ Per Sequence GC Content, chr11.fastq | Рис. 10. ❗ Per Sequence GC Content, chr11_trim.fastq |
| Per Sequence GC Content показывает GC-содержание вдоль всей длины каждого из сиквенсов в файле и сравнивает
его с модельным нормальным распределением (синий график). В случайной библиотеке распределение содержания GC близко к нормальному,
в котором центральный пик соответсвует общему содержанию GC в геноме. Warning выдается в случае, когда суммарные отклолнения от
нормального распределения составляют более 15%, Failure - более 30%. Необычная форма распредления, сильно отклоняющаяся от нормального, чаще всего сигнализирует о загрязненности библиотеки. Расширенные пики свидетельствуют о присутсвии загрязнителей разного рода. Слишком острый пик при более менее гладких остальных частях, обычно появляется при наличии специфического загрязнителя (например димеров адаптеров). Этот случай часто сопряжен с отклонениями в модуле Overrepresented sequences. Такая ситуация, к слову, наблюдается и в нашей последовательности. Как до, так и после чистки пик распределения слишком острый, а программа выдает Warning одновременно в этом модуле и в модуле Overrepresented sequences. Существенных различий между графиками до и после тримминга вновь не видно, хотя можно заметить, что правая часть графика в очищенной последовательности стала чуть более гладкой. |
|
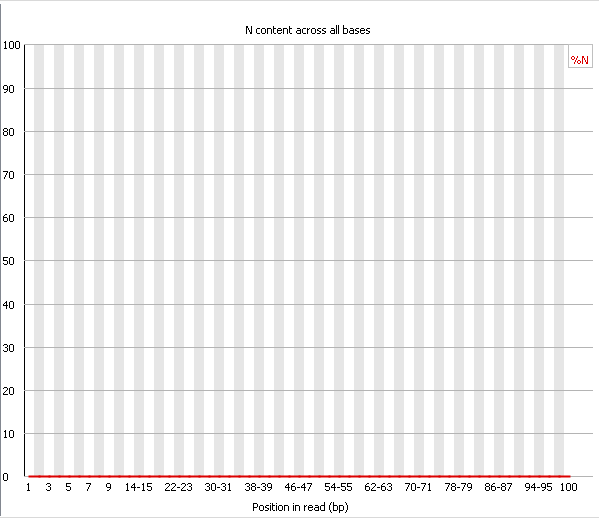 | |
| Рис. 11. ✔ Per Base N Content, chr11.fastq | Рис. 12. ✔ Per Base N Content, chr11_trim.fastq |
| Per Base N Content. Если секвенатор не может распознать основание с достаточной уверенностью, он
заменяет его на символ N. График Per Base N Content показывает процентное содержание N в каждой позиции. Если для какой-либо
позиции N content >5%, выдается Warning, >20% - Failure. Причиной ошибок в этом модуле обычно служит общее низкое качество
проведенного секвенирования. В нашей последовательности как до, так и после чистки, неопределенных оснований не встречается, что, безусловно, очень хорошо. |
|
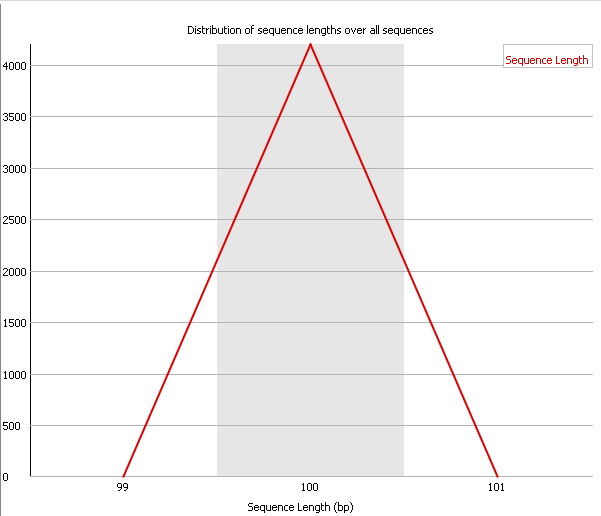 | 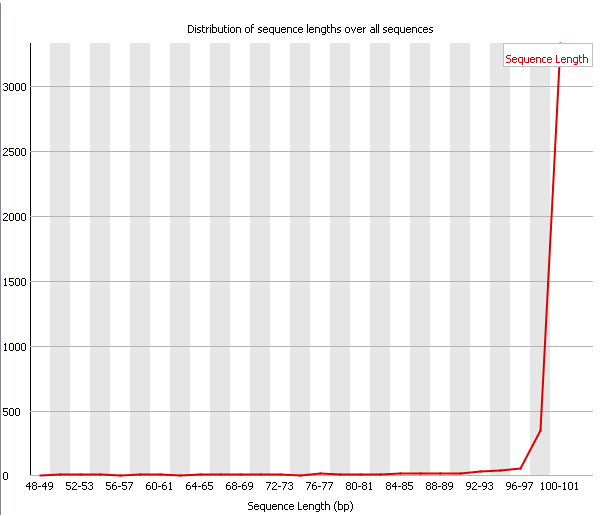 |
| Рис. 13. ✔ Sequence Length Distribution, chr11.fastq | Рис. 14. ❗ Sequence Length Distribution, chr11_trim.fastq |
| Sequence Length Distribution показывает распределение длин анализируемых фрагментов. В норме график
имеет только один пик, соответсвующий наиболее часто встречающейся длине фрагмента. В случае, когда не все фрагменты имеют
одинаковую длину, выдается Warning. Если какой-то фрагмент имеет нулевую длину, выдается Failure. Для некоторых платформ
анализ фрагментов разной длины является нормальным, и Warning в этом модуле может быть проигнорирован. Для нашей последовательности графики до чистки и после на первый взгляд значительно отличаются. Прежде всего, стоит указать, что до чистки анализировались только фрагменты длины 100, а полсе работы Trimmomatic с параметром MINLEN:50 - 50-100. Однако фактически число фрагментов длины меньше 100 очень мало, поэтому плечи графика на рисунке 14 очень пологие и пик наблюдается на той же длине в 100 bp. Для очищенной последовательности программа выдает Warning в виду наличия небольшого числа фрагментов, отличающихся по длине, однако это не повод для беспокойства. |
|
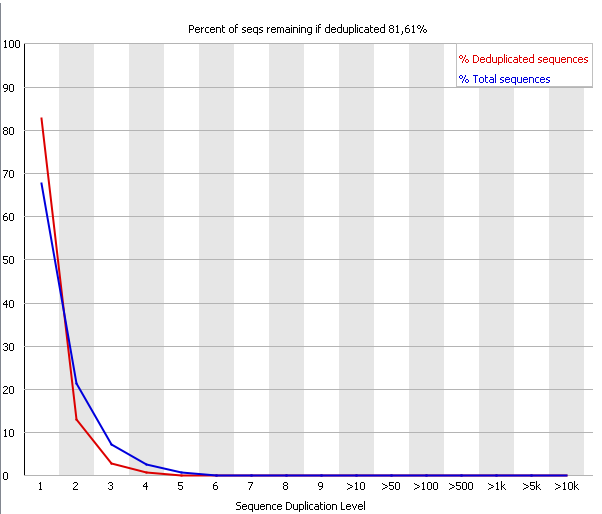 | 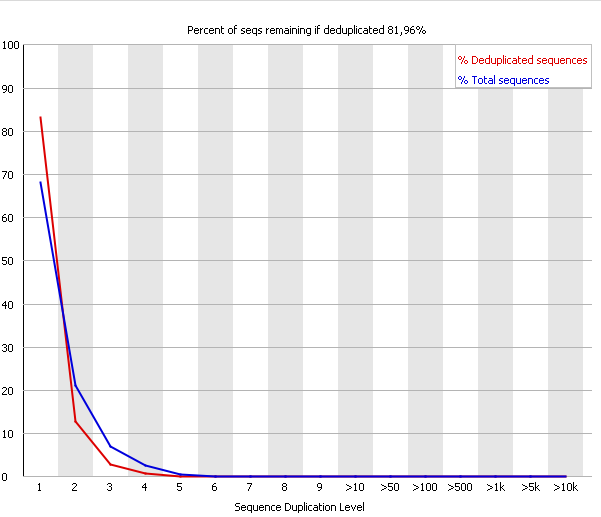 |
| Рис. 15. ✔ Duplicate Sequences, chr11.fastq | Рис. 16. ✔ Duplicate Sequences, chr11_trim.fastq |
| Duplicate Sequences подсчитывает уровень дупликации каждой последовательности библиотеки и создает
график, показывающий относительные количества последовательностей с разными уровнями дупликации. Синяя линия показывает, как
распределены уровни дупликации по всему набору последовательностей. Красная линия - распределение после дедупликации. В хороших
библиотеках показатели для большинства последовательностей смещены в левую часть графика (как для красной, так и для синей линий).
Излишние дупликации по какой-либо последовательности или загрязнение могут приводить к появлению пиков в правой части.
В случае, когда неуникальные последовательности составляют более 20% программа выдает Warning, более 50% - Failure. Для нашей последовательности проблем с этим модулем не возникло. Графики до и после чистки практически идентичны. |
|
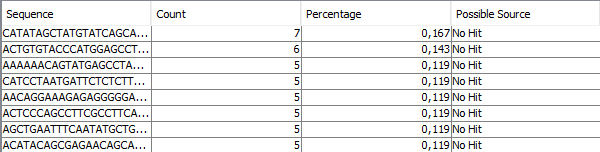 | 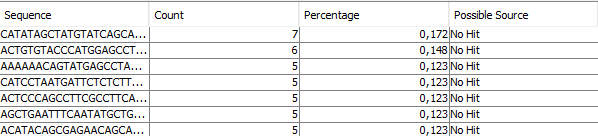 |
| Рис. 17. ❗ Overrepresented Sequences, chr11.fastq | Рис. 18. ❗ Overrepresented Sequences, chr11_trim.fastq |
| Overrepresented Sequences. В нормальной разнообразной библиотеке, как правило, содержится много
различных последовательностей, ни одна из которых не составляет значительной части от целого. Если какая-то последовательность
представлена в слишком большом количестве, это сигнализирует либо о ее высокой биологической значимости, либо о загрязнении библиотеки.
В данном модуле перечисляются все последовательности, составляющие более 0.1% от целого. Каждую из таких последовательностей программа
также прогоняет по базе наиболее распространенных загрязнителей и выдает наиболее близкое совпадение (hit), если такое имеется.
При наличии хотя бы одной последоваетльности, встречающейся чаще 0.1%, выдается Warning, при наличии последовательности, встречающейся
чаще 1% - Failure. В нашей последовательности overepresented sequences встречаются, FastQC выдает предупреждение. Возможный источник программой не определяется. После чистки количество overepresented sequencesуменьшается на 1 (с 8 до 7). |
|
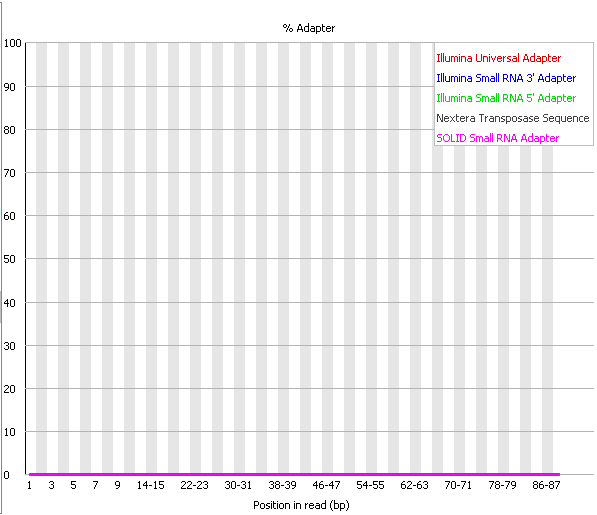 | |
| Рис. 19. ✔ Adapter Content, chr11.fastq | Рис. 20. ✔ Adapter Content, chr11_trim.fastq |
| Adapter Content показывает суммарное процентное содержание частей библиотеки, в которых была найдена
последовательность адаптера в какой-либо из позиций. Как только последовательноять адаптера найдена в риде, она считается присутствующей
до самого конца, поэтому процент будет только увеличиваться по мере продвижения по всей длине. Если последовательность присутствует
более, чем в 5% ридов, выдается Warning, 10% - Failure. В нашей последовательности адаптеры уже были удалены, поэтому как до, так и после чистки график представляет собой прямую линиию в районе нуля. |
|
Картирование чтений
Чтения были откартированы с помощью программы BWA - Burrows-Wheeler Alignment Tool.
Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| bwa index chr11.fasta | Индексирование референсной последовательности | Индексированный файл chr11.fasta |
| bwa mem chr11.fasta chr11_trim.fastq > chr11.sam | Выравнивание очищенных чтений с референсной последовательностью | Файл chr11.sam, содержащий выравнивание в формате SAM (Sequence Alignment/Map) |
Анализ выравнивания
Для анализа полученного выравнивания применялась программа Samtools,
предназначенная для обработки файлов в формате SAM.
Использованные команды указаны в таблице.
| Команда | Назначение | Результат |
|---|---|---|
| samtools view chr11.sam -b -o chr11.bam | Перевод выравнивания в бинарный формат .bam (опция -b меняет формат выходного файла с установленного по умолчанию, -o обозначат имя выходного файла). | chr11.bam |
| samtools sort chr11.bam -T smth.txt -o chr11_sort.bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения (опция -T позволяет записывать временные файлы в файл smth.txt, а не в stdout). | chr11_sort.bam |
| samtools index chr11_sort.bam | Индексирование отсортированного выравнивания | Индексированный chr11_sort.bam |
| samtools idxstats chr11_sort.bam > reads.out | Выяснение числа чтений, откартированных на геном | reads.out |
Из файла reads.out, я получила информацию, что на хромосому откартировалось 4062 чтения и 2 чтения откартированы не были.
Определение среднего покрытия экзона
Для начала было вычислено покрытие для каждого из нуклеотидов и был выбран один из нуклеотидов с наилучшим покрытием (185). Он имел координату 116628513. Данный нуклеотид был найден в Genome Browser. Выяснилось, что он принадлежит гену BUD13 c координатами complement(116618886..116643714) и длиной 24829. Экзон, содержащий данный нуклеотид, имеет границы 116628481-116628666.
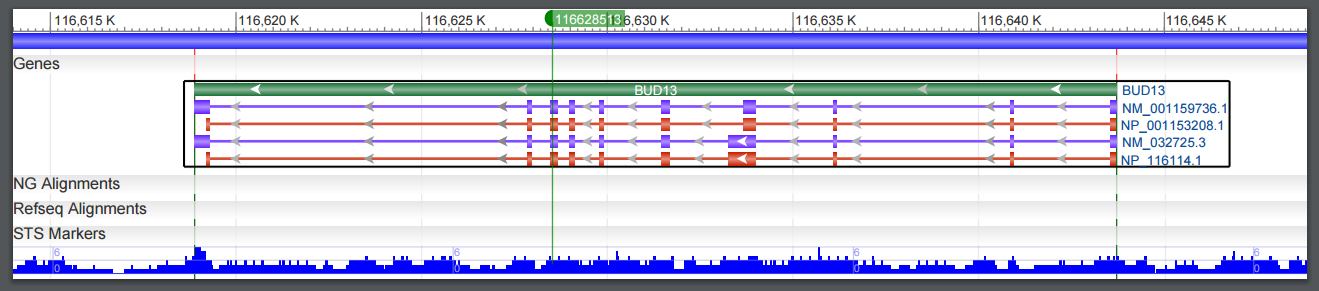 |
| Рис. 21. Ген BUD13 |
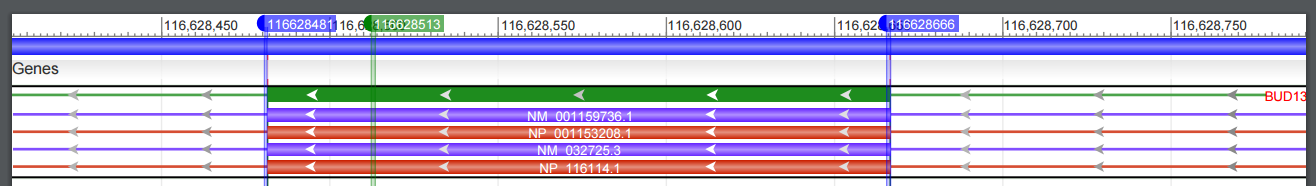 |
| Рис. 22. Экзон, содержащий нуклеотид 116628513 |
Использованные команды:
| Команда | Назначение | Результат |
|---|---|---|
| samtools depth chr11_sort.bam > depth.out | Вычисление покрытия каждого нуклеотида | depth.out |
| samtools depth -r chr11:116628481-116628666 chr11_sort.bam > exon.out | Вычисление покрытия нуклеотидов в границах экзона | exon.out |
Исходя из данных файла exon.out с помощью Excel было вычислено среднее покрытие выбранного экзона. Оно составило 143.48, что является достаточно хорошим показателем.
Поиск SNP и инделей
| Команда | Назначение | Результат |
|---|---|---|
| samtools mpileup -uf chr11.fasta chr11_sort.bam -o snp.bcf | Создание файла с полиморфизмами в формате .bcf | snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf | snp.vcf |
Всего в файле snp.vcf указано 23 полиморфизма, из них 1 вставка, 1 делеция и 21 замена. Покрытие и качество полиморфизмов встречается как хорошее (например качество 188.009 и покрытие 92), так и не очень (например качество 4.76921 и покрытие 2). Среднее значение качества - 118,82, покрытия - 23,56. В целом показатели достаточно нормальные.
| Примеры полиморфизмов | |||||
|---|---|---|---|---|---|
| Координата | Тип | В референсе | В прочтении | Качество прочтения на участке | Глубина покрытия на участке |
| 116628401 | Замена | T | С | 188.009 | 92 |
| 116650454 | Делеция | ATCTCT | ATCT | 196.468 | 22 |
| 116657590 | Вставка | CAAA | CAAAA | 144.467 | 63 |
Аннотация SNP
Далее необходимо было аннотировать полученные SNP с помощью программы ANNOVAR. Для этого сначала нужно было подготовить входной файл, чтобы программа моглас ним работать. В первую очередь из файла snp.vcf были удалены все индели, полученный файл - snp_noindel.vcf. Затем был использован скрипт convert2annovar.pl.
| Команда | Назначение | Результат |
|---|---|---|
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_noindel.vcf -outfile snp.avinput | Подготовка входного файла | snp.avinput |
Полученный файл использовался для аннотации SNP по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar c помощью скрипта annotate_variation.pl.
В Annovar существуют 3 типа аннотаций по базам данных, основанных на: генной разметке (gene-based annotation); разметке других регионов генома (region-based annotation); фильтрации (filter-based annotation). Тип использованной аннотации указан для каждой базы данных отдельно.
Refgene
Тип аннотации: gene-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr11.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
- chr11.refgene.variant_function - содержит описание всех полиморфизмов.
- chr11.refgene.exonic_variant_function - содержит описание полиморфизмов внутри экзонов.
- chr11.refgene.log - содержит отчет о работе команды.
База данных refseq в annovar делит snp на несколько категорий, которые отображены в первой колонке файла chr11.refgene.variant_function. Они указывают, где находится данный полиморфизм - внутри экзона, интрона, гена некодирующей РНК и т.п.
Возможные категории:
- exonic - полиморфизм внутри экзона (частично или полностью)
- splicing - полиморфизм в пределах 2 bp от границы сплайсинга (число bp можно изменить)
- ncRNA - полиморфизм полностью или частично входит в транскрипт, не имеющий аннотации как кодирующий
- UTR5 - полиморфизм полностью или частично входит в 5′-нетранслируемую область
- UTR3 - полиморфизм полностью или частично входит в 3′-нетранслируемую область
- intronic - полиморфизм полностью или частично внутри интрона
- downstream - полиморфизм в пределах 1-kb downstream от сайта окончания транскрипции (параметр может быть изменен)
- upstream - полиморфизм в пределах 1-kb upstream от сайта начала транскрипции (параметр может быть изменен)
- intergenic - полиморфизм на пересечении генов
Распределение SNP по группам в моем файле:
| exonic | intronic | UTR3 | Всего |
|---|---|---|---|
| 5 | 12 | 4 | 21 |
13 полиморфизмов являются гетерозиготными, 8 - гомозиготными.
Если полиморфизм попадает в категории exonic/intronic/ncRNA, то во второй колонке того же файла указывается имя соответсвующего гена (если это сразу несколько генов, то они разделяются запятой). Если полиморфизм попадат в какую-либо другую категорию, то во второй колонке будут указаны два соседних гена и расстояние между ними.
В моем случае в категории, для которых указывается ген, попали 17 полиморфизмов. Все они расположены на одном из трех генов - KCNJ11, BUD13 или ZPR1.
Ген KCNJ11 кодирует белок потенциалозависимого калиевого ионного канала. В нем оказалось 2 полиморфизма категории exonic.
Ген BUD13 является белок кодирующим, однако функция его в NCBI не прописана. В нем оказалось 7 полиморфизмов категории intronic и 2 - exonic.
Ген ZPR1 кодирует белок, который взаимодействует с белком выживания двигательных нейронов (survival motor neuron protein - SMN1), усиливая сплайсинг пре-мРНК и активируя нейронную дифференцировку и рост аксонов. В нем оказалось 5 полиморфизмов категории intronic и 1 полиморфизм категории exonic.
Большинство аннотированных полиморфизмов (57%) принадлежат категории intronic, и лишь 5 (23%) - категории exonic. Это можно считать достаточно логичным, так как замены в некодирующих последовательностях не подвергаются такому тщательному отбору, как замены в последовательностях, кодирующих белки. Полиморфизмы категории exonic могут приводить к аминокислотным заменам и, в случае несинонимичных замен, к нарушению функии белка.
В таблице более подробно рассмотрены полиморфизмы, попавшие внутрь экзонов, с указанием нуклеотидных и аминокислотных замен:
| № | Координаты (*) | Ген | Н. замена | Тип замены | Синонимичность | Качество чтений | Глубина покрытия | А.к. замена |
|---|---|---|---|---|---|---|---|---|
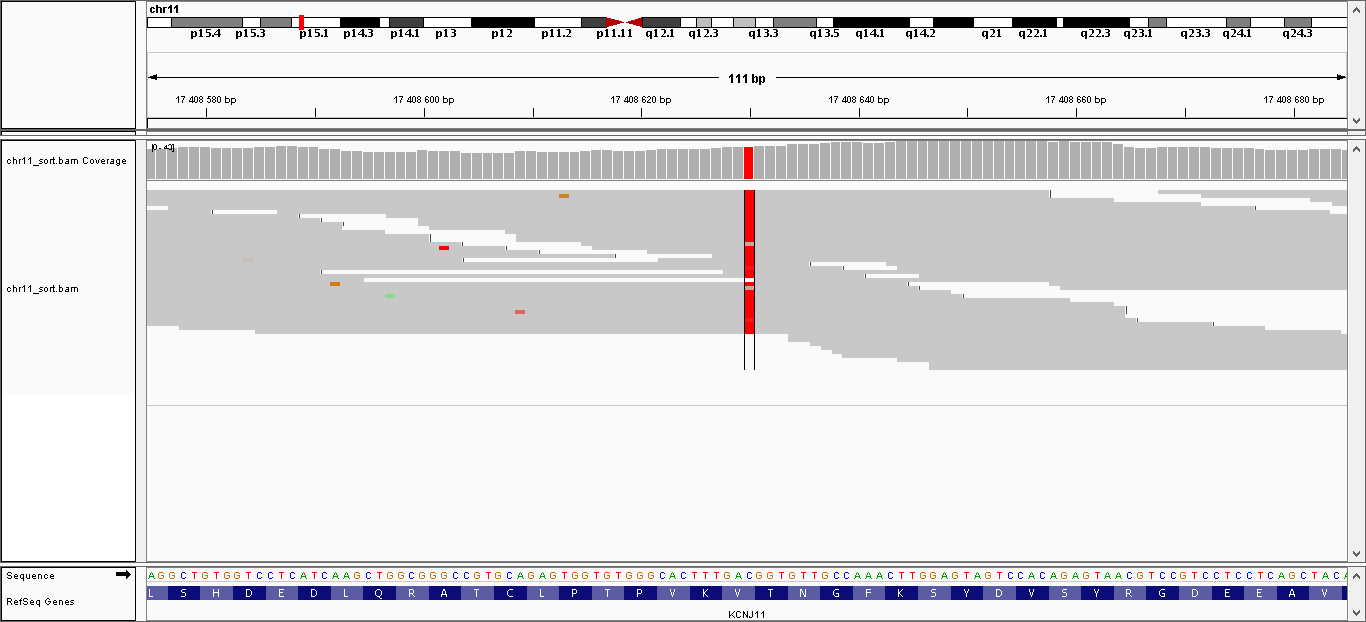
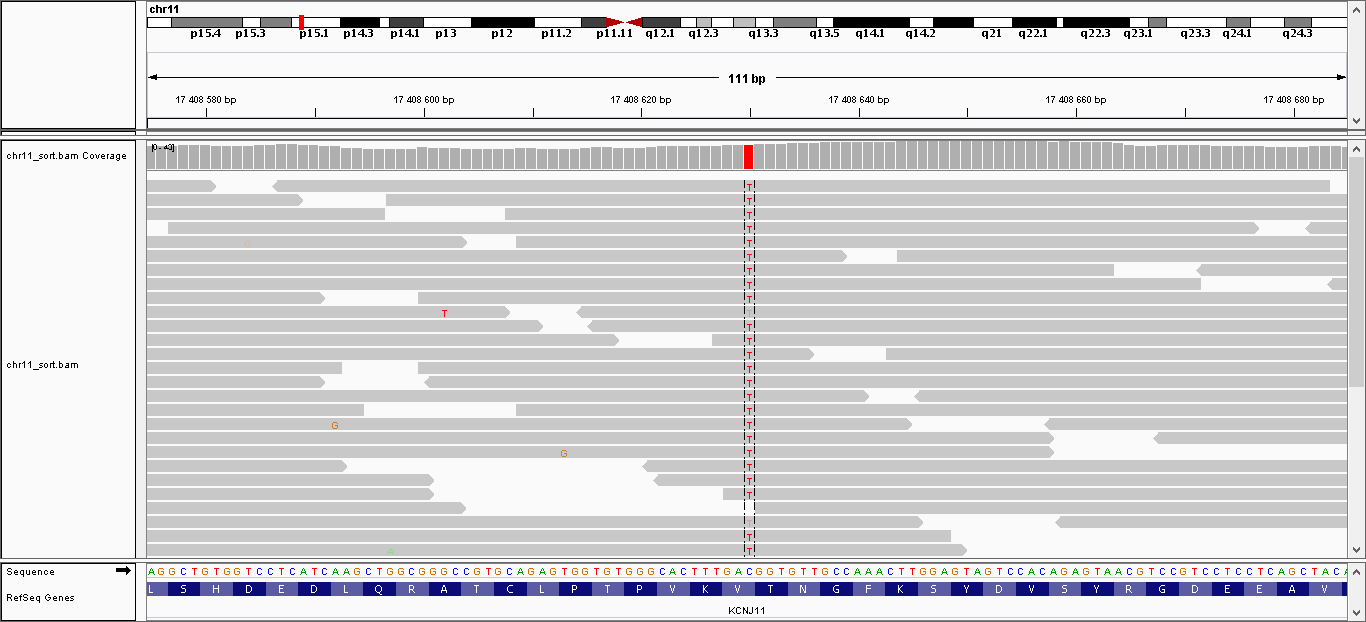
| 1 | 17408630 | KCNJ11 | C > T | гомозиготная | нет | 221,999 | 35 | V250I, V337I |
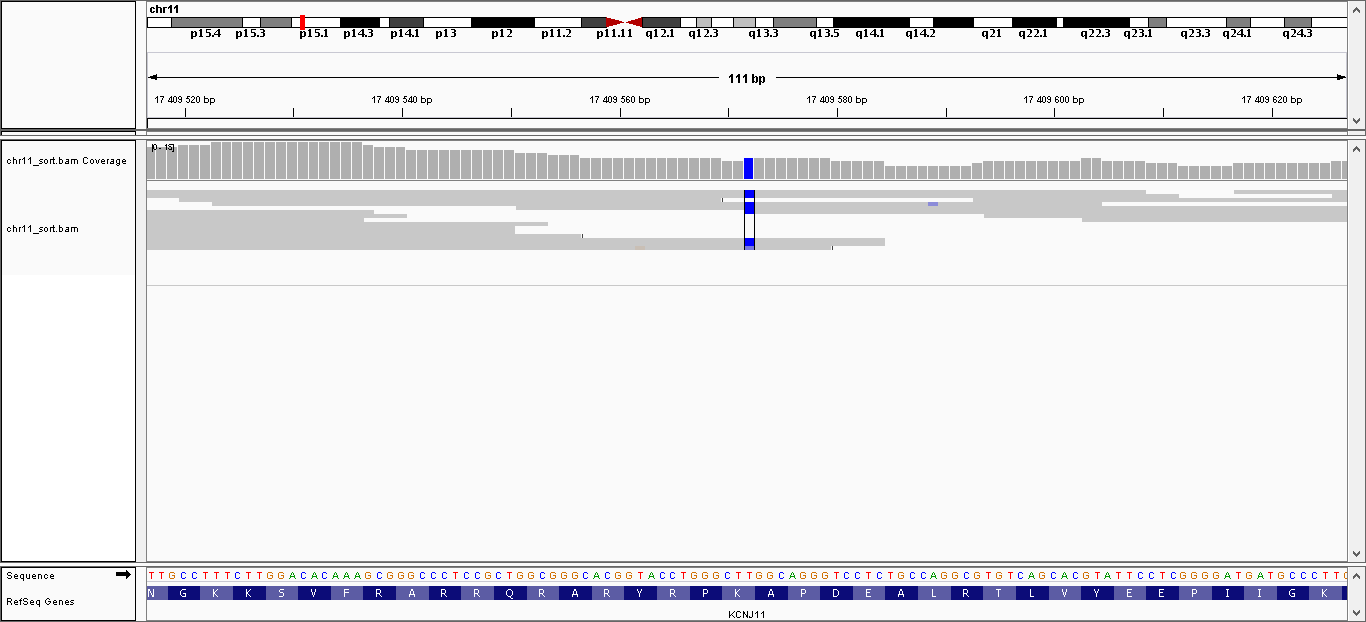
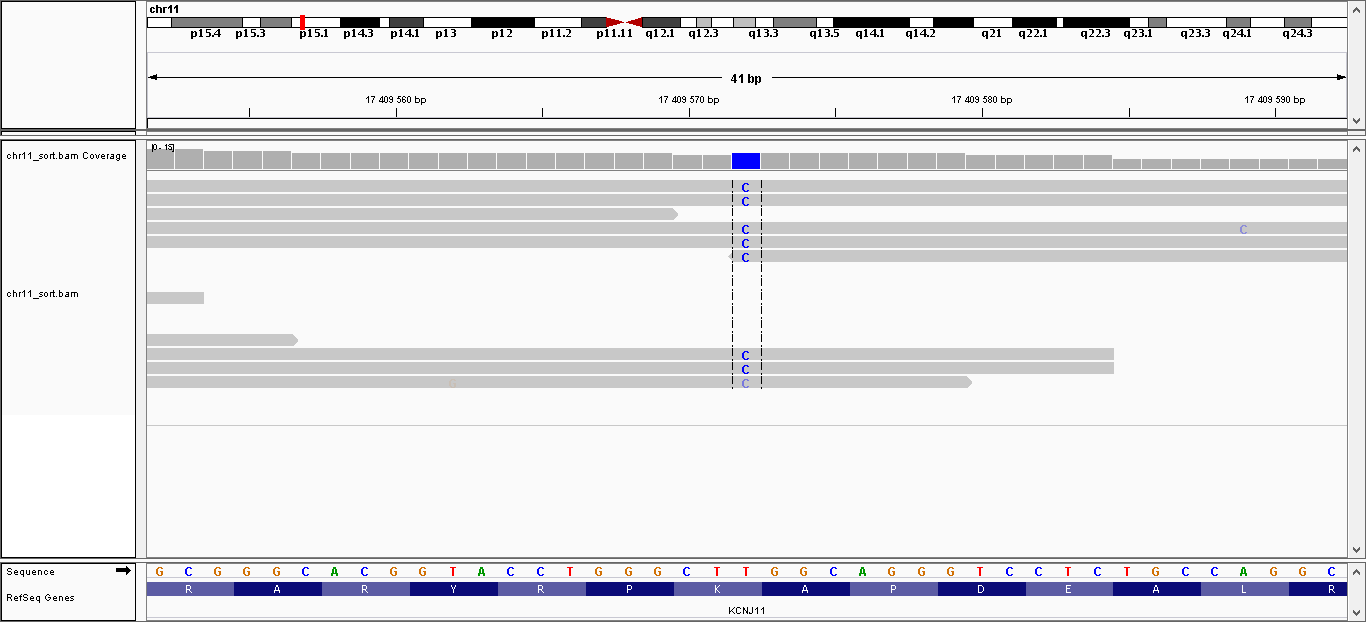
| 2 | 17409572 | KCNJ11 | T > C | гомозиготная | нет | 138,133 | 8 | K23E |
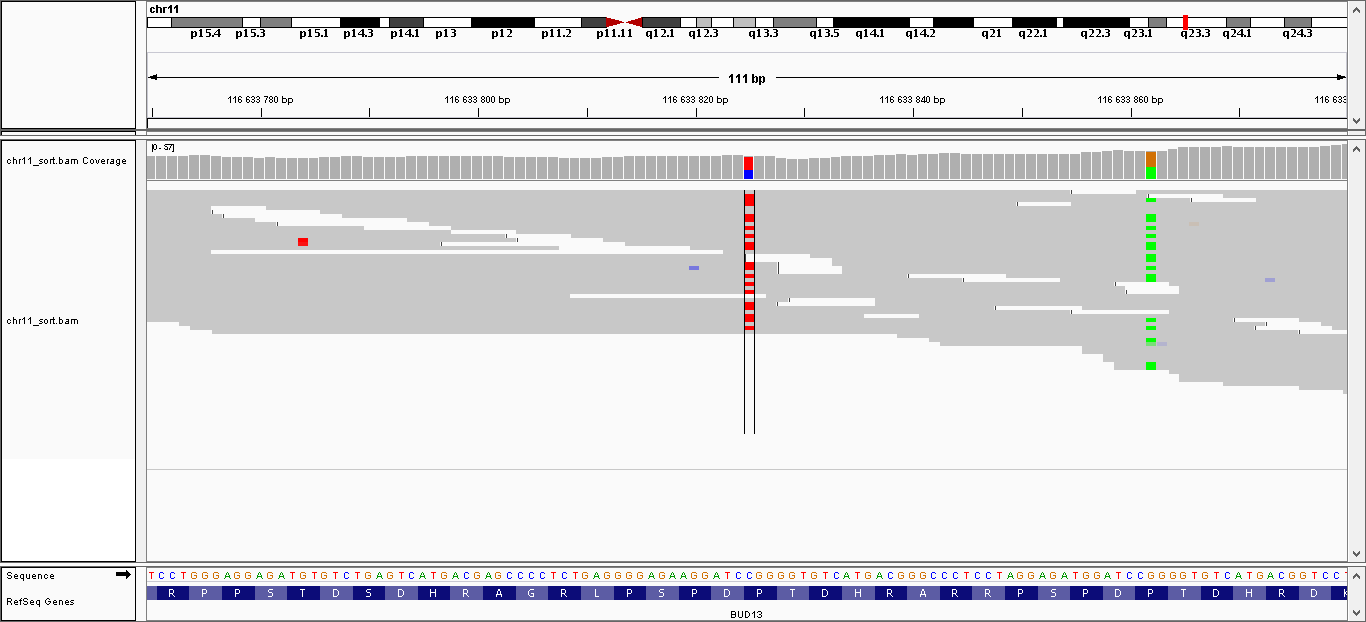
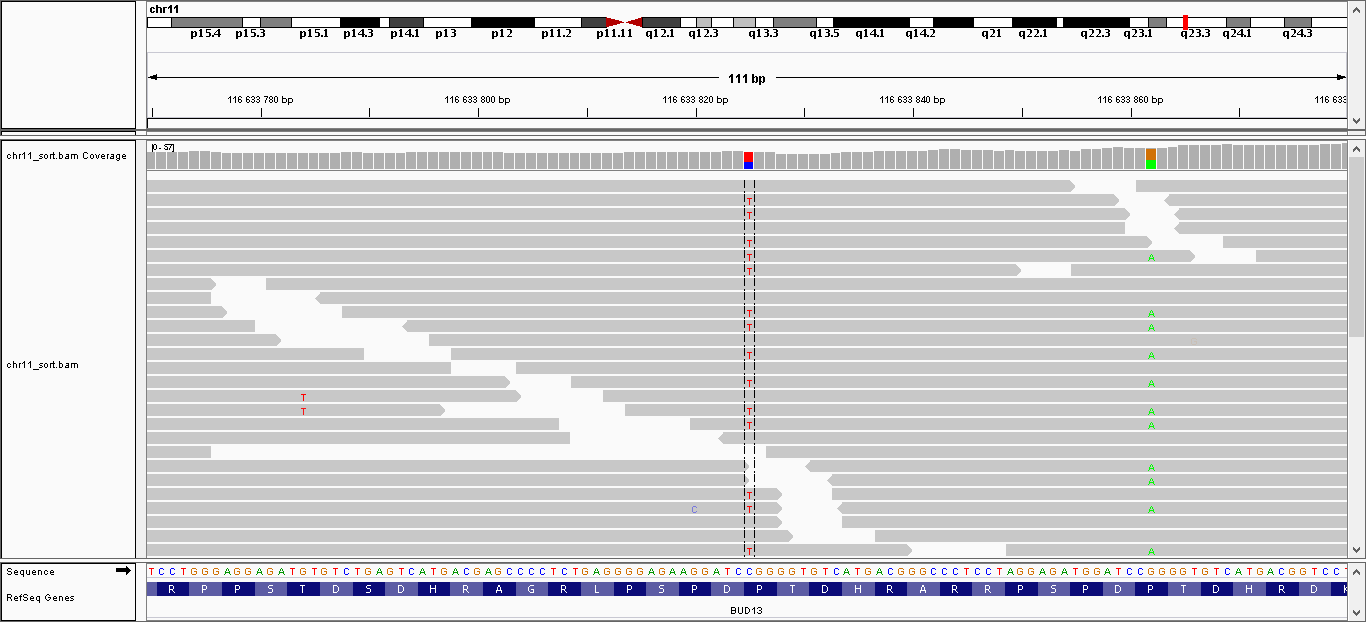
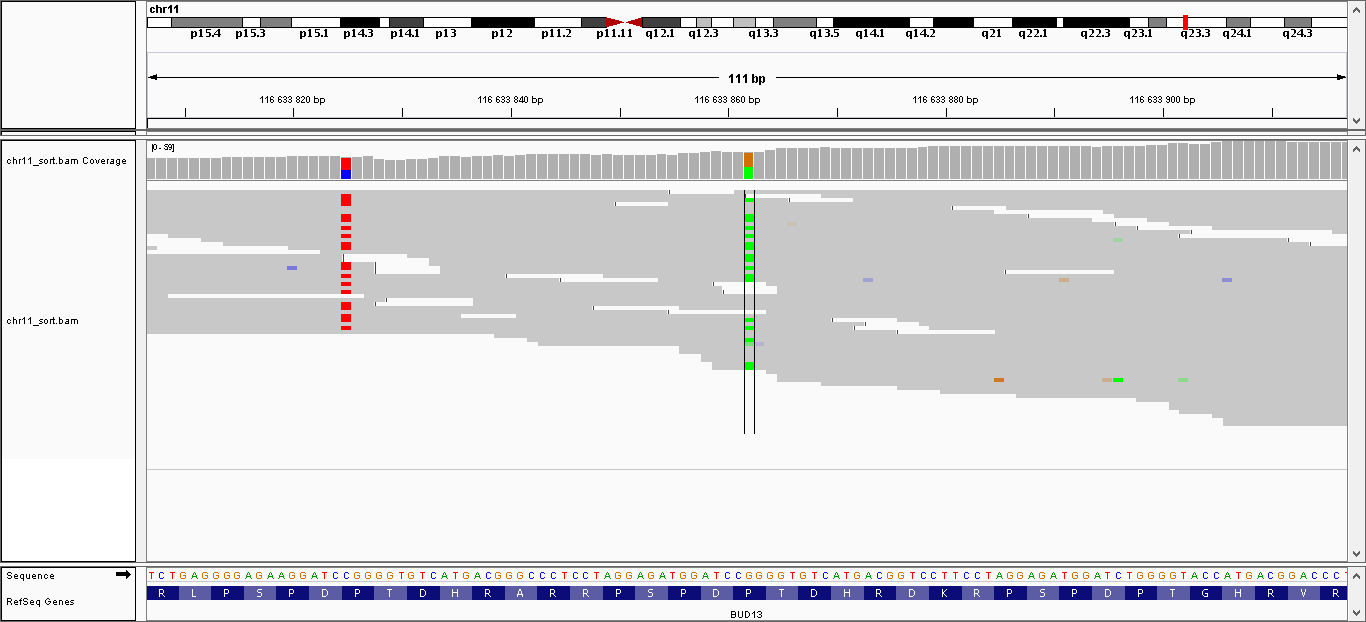
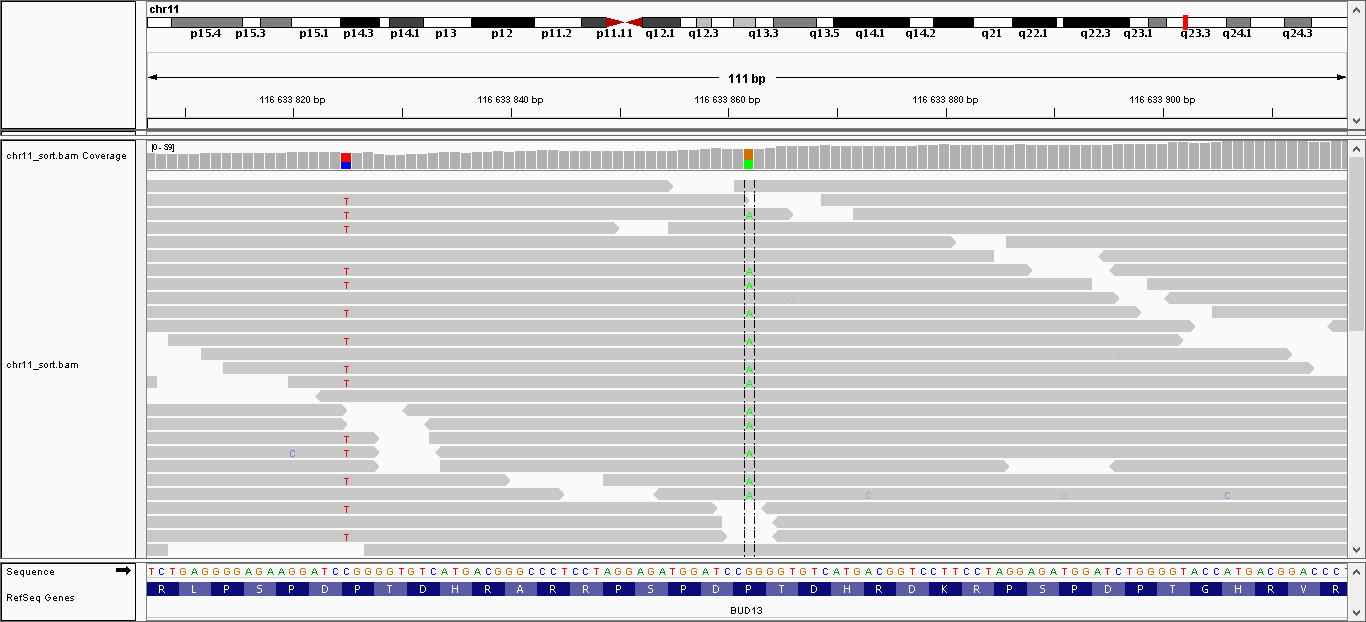
| 3 | 116633825 | BUD13 | C > T | гетерозиготная | да | 194,009 | 33 | P160P |
| 4 | 116633862 | BUD13 | G > A | гетерозиготная | нет | 224,009 | 40 | P148L |
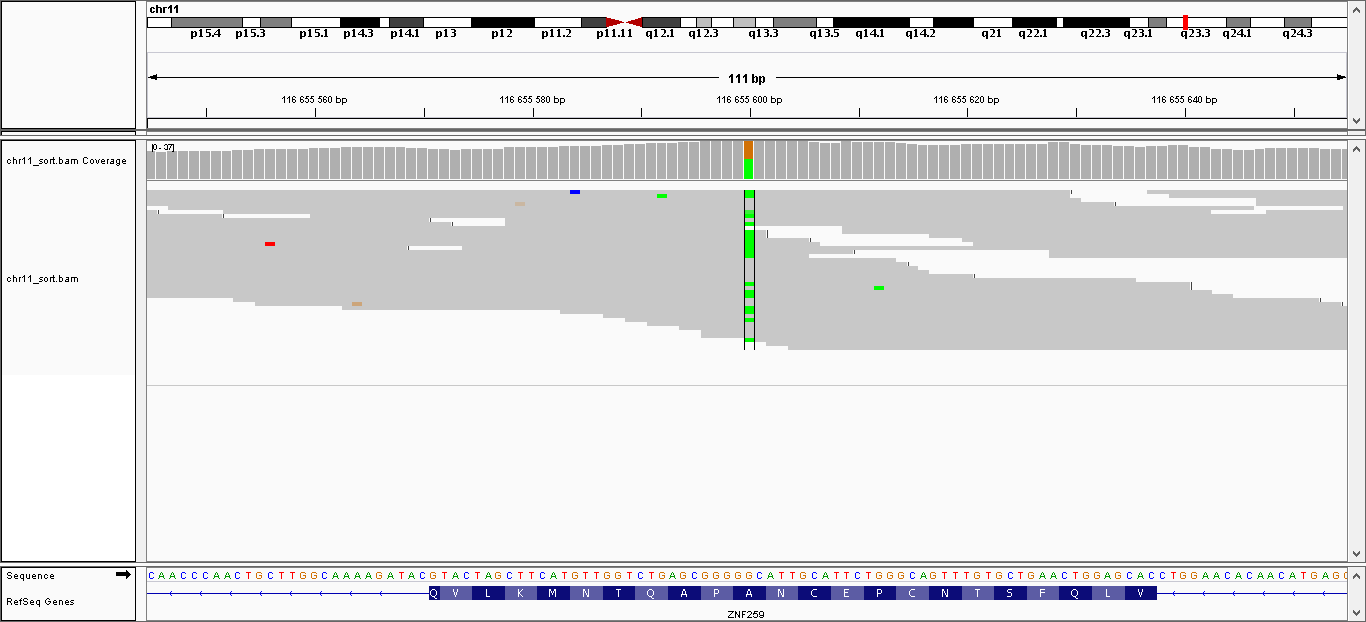
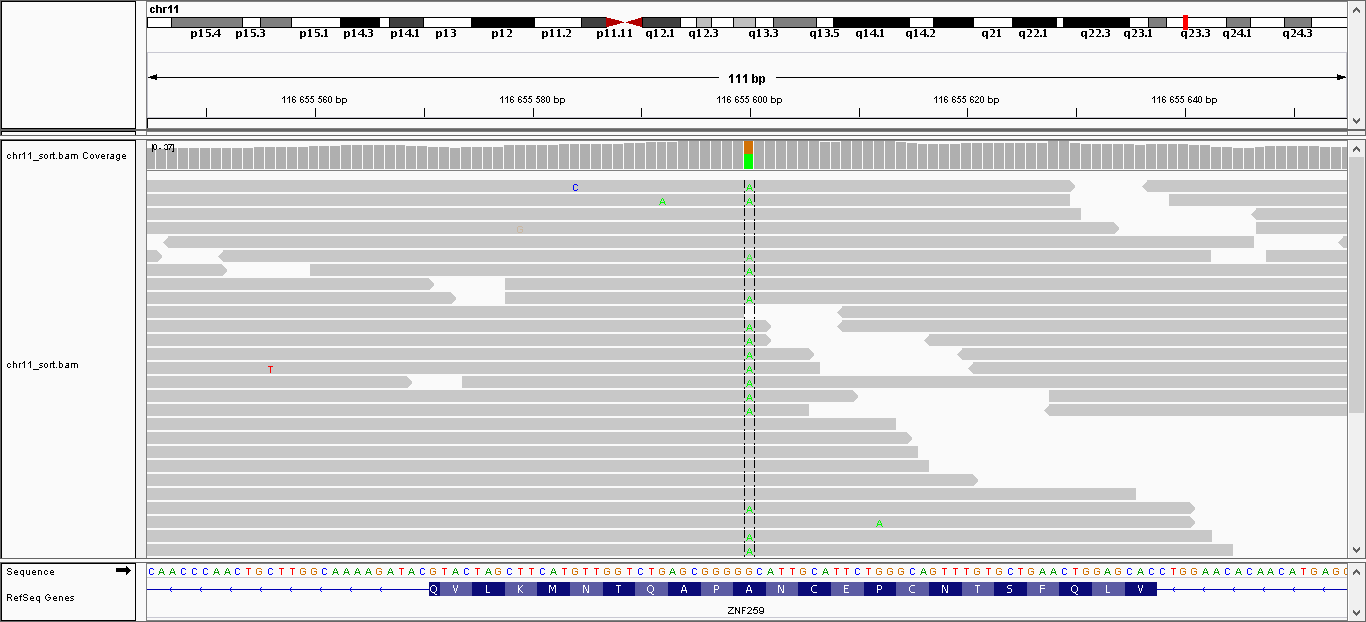
| 5 | 116655600 | ZPR1 | G > A | гетерозиготная | нет | 225,009 | 37 | A264V |
Данные полиморфизмы были визуализированы с помощью программы IGV. Полученные изображения можно увидеть, пройдя по ссылкам:
- Полиморфизм 1: общий вид, увеличение
- Полиморфизм 2: общий вид, увеличение
- Полиморфизм 3: общий вид, увеличение
- Полиморфизм 4: общий вид, увеличение
- Полиморфизм 5: общий вид, увеличение
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dbsnp
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.dbsnp -build hg19 -dbtype snp138 snp.avinput
/nfs/srv/databases/annovar/humandb/
Полученнные файлы:
- chr11.dbsnp.hg19_snp138_dropped - содержит полиморфизмы, имеющие rs в dbsnp, то есть аннотированные в этой базе данных.
- chr11.dbsnp.hg19_snp138_filtered - содержит отфильтрованные полиморфизмы (не имеющие rs в dbsnp).
- chr11.dbsnp.log - содержит отчет о работе команды.
Аннотация по базе dbsnp показала, что 19 полиморфизмов имеют rs, а 2 не имеют. При этом можно заметить, что полиморфизмы, не имеющие rs, обладают достаточно низким качеством (4.76921 и 9.52546) и глубиной покрытия (2 и 1), в то время как аннотированные snp демонстрируют хоть и разные, но в целом лучшие показатели.
1000 genomes
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out
chr11.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
- chr11.1000.hg19_ALL.sites.2014_10_dropped - содержит полиморфизмы, имеющие rs в 1000 genomes, и их частоты.
- chr11.1000.hg19_ALL.sites.2014_10_filtered - содержит полиморфизмы, не имеющие rs в 1000 genomes.
- chr11.1000.log - содержит отчет о работе команды.
Аннотация по 1000 genomes привела к схожему результату. 19 snp по-прежнему имеют rs, а 2 не имеют, ситуация с качеством и глубиной прочтения та же. Дополнительно мы смогли узнать частоты аннотированных полиморфизмов. Они варьируют от 0.00339457 до 0.860623. Средний показатель - 0,34685563.
| № | Координата SNP | Ген | Частота |
|---|---|---|---|
| 1 | 17408630 | KCNJ11 | 0,730631 |
| 2 | 17409572 | KCNJ11 | 0,737021 |
| 3 | 116633825 | BUD13 | 0,545128 |
| 4 | 116633862 | BUD13 | 0,088858 |
| 5 | 116655600 | ZPR1 | 0,046725 |
Видно, что найденные нами экзонные полиморфизмы встречаются достаточно часто, для 3 из 5 snp вероятность превышает 50%.
Gwas
Тип аннотации: region-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr11.gwas
-build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
- chr11.gwas.hg19_gwasCatalog - содержит snp, имеющие известное клиническое значение.
- chr11.gwas.log - содержит отчет о работе команды.
Аннотация по Gwas показала, что из исследуемых нами полиморфизмов 6 имеют известное клиническое значение. Два связаны с развитием диабета 2-го типа, два - с триглицеридами и кровяным давлением, один - с метаболическим синдромом и один - с качествами, ассоциированными с ожирением.
| № | Координата | Ген | Клиническое значение |
|---|---|---|---|
| 1 | 17408630 | KCNJ11 | Диабет 2-го типа |
| 2 | 17409572 | KCNJ11 | Диабет 2-го типа |
| 4 | 116633862 | BUD13 | Метаболический синдром |
Clinvar
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out
chr11.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
- chr11.clincar.hg19_clinvar_20150629_dropped - содержит snp, аннотированные в Clinvar.
- chr11.clincar.hg19_clinvar_20150629_filtered - содержит snp, не аннотированные в Clinvar.
- chr11.clincar.log - содержит отчет о работе команды.
В Clinvar оказались аннотированы только 2 snp и, соответсвенно, 19 оказались не аннотированы. Оба аннотированных snp являются экзонными. В таблице они рассмотрены более подробно.
| № | Координата | Ген | Клиническая значимость | Связанные заболевания и состояния |
|---|---|---|---|---|
| 1 | 17408630 | KCNJ11 | Не патогенна | Не указано |
| 2 | 17409572 | KCNJ11 | Восприимчивость к лекарственным препаратам; не патогенна | Сахарный диабет 2-го типа; стрессовый ответ при физической нагрузке; перманентный сахарный диабет новорождённых |
Сводная таблица SNP
Сводную таблицу Excel можно скачать по ссылке.
Использовались только файлы с анноированными snp (dropped).
Сводная таблица со всеми командами в блоке
| № | Команда | Назначение |
|---|---|---|
| 1 | fastqc chr11.fastq | Анализ качества чтений исходной последовательности |
| 2 | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr11.fastq chr11_trim.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| 3 | fastqc chr11_trim.fastq | Анализ качества чтений очищенной последовательности |
| 4 | bwa index chr11.fasta | Индексирование референсной последовательности |
| 5 | bwa mem chr11.fasta chr11_trim.fastq > chr11.sam | Выравнивание очищенных чтений с референсной последовательностью |
| 6 | samtools view chr11.sam -b -o chr11.bam | Перевод выравнивания в бинарный формат .bam |
| 7 | samtools sort chr11.bam -T smth.txt -o chr11_sort.bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения |
| 8 | samtools index chr11_sort.bam | Индексирование отсортированного выравнивания |
| 9 | samtools idxstats chr11_sort.bam > reads.out | Выяснение числа чтений, откартированных на геном |
| 10 | samtools depth chr11_sort.bam > depth.out | Вычисление покрытия каждого нуклеотида |
| 11 | samtools depth -r chr11:116628481-116628666 chr11_sort.bam > exon.out | Вычисление покрытия нуклеотидов в границах экзона |
| 12 | samtools mpileup -uf chr11.fasta chr11_sort.bam -o snp.bcf | Создание файла с полиморфизмами в формате .bcf |
| 13 | bcftools call -cv snp.bcf -o snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf |
| 14 | perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_noindel.vcf -outfile snp.avinput | Подготовка входного файла для ANNOVAR |
| 15 | perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr11.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Refgene |
| 16 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Dbsnp |
| 17 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в 1000 genomes |
| 18 | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr11.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Gwas |
| 19 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr11.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Clinvar |