AN ATLAS OF CONTACTS OF A Saccharomyces cerevisiae
ALCOHOL DEHYDROGENASE I |
RESUMES
This page is dedicated to the compilation of the atlas of contacts of alcochol dehydrogenase I
is the constitutive enzyme of yeast (Saccharomyces cerevisiae) that reduces acetaldehyde to ethanol
during the fermentation of glucose
[1] .
The structure of the atlas includes the description:
- The low molecular weight ligands in the complex
- Protein-protein contacts (covalent bonds, hydrogen bonds, salt bridges, hydrophobic core)
- The ligand-biomolecule contacts
The information about all contacts is accompained by graphic illustrations obtained by using JMol program.
The images are available as Jmol-applets. On the links, you can go to the appropriate scripts.
If applets do not work, to get acquainted with them, you can click on the link :
http://kodomo.fbb.msu.ru/~polina.shpudeiko/term2/atla..
Read an article in Russian
|
INTRODUCTION |
|
Discussed in this study, the complex is found in baker's yeast(Saccharomyces cerevisiae strain ATCC
204508 / S288c)[2], commonly used
in the manufacture of bakery and alcohol products, and used as a popular model in various scientific studies
[3].
Alcohol dehydrogenase I involved in the process of fermentation Glucose: converts acetaldehyde into ethanol
(Fig.1)[1].
This isozyme catalyzes the reaction of converting primary alcohols to the corresponding unbranched
aldehydes and it is also active against secondary alcohols. Its catalytic activity can be illustrated by the following reaction
:
Alcochol + NAD + <=> aldehyde or ketone + NADH[3]
Alcohol dehydrogenase I refers to the class of oxidoreductases [1]
(enzymes which catalyze reactions of biological oxidation, accompanied by transfer of electrons from one molecule to another
[4]),
is homotetramer (protein complex consisting of four identical subunits associated non-covalently
[5]).
Four different protein subunits arranged in similar dimer called AB and CD. Subunit A and C, arranged in various
dimers structurally similar (similar to each other B and D), however, between the structure of the two pairs, there are
some differences, which form dimers asymmetry in the structure.
Probably, this asymmetry plays an important role in the catalytic mechanism of the enzyme
[1].
The complex also includes low molecular weight ligands: trifluoroethanol, zinc ions and NAD. More details
are available for consultation in the section, devoted to the description of the low molecular weight ligands
(see below)
[1].
General information about the alcohol dehydrogenase I compiled in table 1.
|
Table 1. General information[1]
| The name of protein | Alcohol dehydrogenase 1 |
| ID of the database PDB (PDB ID) | 4W6Z |
| ID of the database Uniprot (Uniprot ID ) | P00330 (ADH1_YEAST) |
| Organism | Saccharomyces cerevisiae |
| Type enzyme | Oxidoreductase |
| The total weight of the structure | 149543,72 |
| The number of subunits in the protein composition | 4 |
| Subunits length (in amino acid residues) | 347 |
| Structure subunits | Homotetramer |
| The name of gene | ADH1 |
| Location in the cell | Cytoplasm |
| Fig.1 The scheme of the fermentation[6] 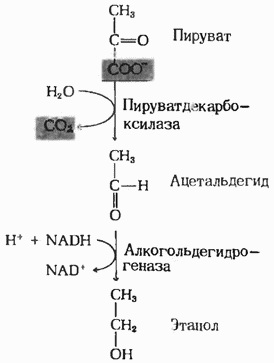 |
|
| |
LOW WEIGHT LIGANDS
The alcohol dehydrogenase 1 consists of three types of ligands:
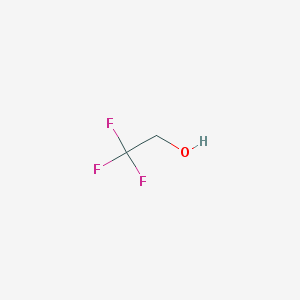
Trifluoroethanol
One molecule located on each of the four chains, however, chains A and C connect with ligand in different way
from similar ligand molecules with chains B and D (see section about ligand-biomolecule contacts). Zinc ion
The structure of the protein complex comprises eight zinc ions, two for each subunit.
The asymmetry in the structure of the dimers' subunits observed in the bonds between the protein chains and trifluoroethanol,
evident in relations zinc ions with the protein chains of a dimer. Thus asymmetries in the structure of two dimers
aren't observed.
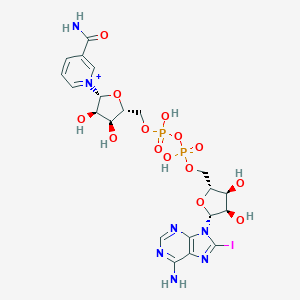
Nicotinamide-8-iodo-adenine-dinucleotide
The protein complex consists of two molecules of the compound,
they are symmetrically arranged on the chains A and C.
The information about the physico-chemical properties of the ligands represented in Table 2.
| THE NAME OF LIGANDS (BY IUPAC) | 2,2,2-Trifluoroethanol | Zinc ion | Nicotinamide-8-iodo-adenine-dinucleotide |
| CHEMICAL FORMULA | C2H3F3O | Zn2+ | C21H27IN7O14P2 |
| MOLECULAR WEIGHT (g/mol) | 100,04 | 65,38 | 790.334 |
| PubChem СID | 6409(PubChem) | 32051(PubChem) | 9543524(PubChem) |
|  |  |  |
|
| |
|
PROTEIN-PROTEIN CONTACTS
The hydrophobic core.
When laying the polypeptide chain of the protein tends to become energetically favorable form, characterized by the
minimum of free energy. Therefore, the hydrophobic amino acid radicals seek to unite within the globular structure of
water-soluble proteins. Between them there are so-called hydrophobic interactions, and van der Waals forces between
closely adjacent to each other atoms. As a result, the hydrophobic core is formed inside the protein globule.
Hydrophilic group of the peptide backbone in the formation of secondary structures form a plurality of hydrogen bonds,
thereby avoiding binding and destruction of their internal water, dense structure of the protein.
This protein complex there are 2 large identical hydrophobic core, located on the neighboring dimers.
The hydrophobic core permeate almost all dimer and sufficiently extensive part of coming to the surface. Thus NAD
partially located in the hydrophobic core, while the other ligand is almost completely immersed in it. From this,
it can be assumed that the hydrophobic core may have a role in the protein binding to the substrate, but probably is
a large hydrophobic core it is very important to maintain the dimer structure, in particular the protein-protein
interaction and maintaining the dimer structure and possibly binding NAD
(see. ligand biomolecular interactions).
In addition, the protein also contains 19 hydrophobic smaller nuclei, the largest of which consists of 43 atoms.
None of these nuclei is not associated with ligands and generally in their distribution observed for this inherent
symmetry between homotetramer dimers. However, the largest of these small nuclei located on the chain C, significantly
higher than the same on the symmetrical chain A.
The total number of atoms in the hydrophobic core - 2782, when the total number of atoms equal to 10601.
That is approximately 26%. That is about a quarter of all the atoms are part of the hydrophobic core. However,
given that most large nucleus permeates almost all of the protein, it can be concluded that the atoms that make up
the nuclei located quite loosely.
- Covalent bond - chemical bond formed by overlapping a pair of valence electron clouds. Provides
the links electron clouds are called total electron pair
[7].
Disulfide bridges- Covalent bond between the two sulfur atoms (-S-S-), members of the sulfur
containing amino acid cysteine. Disulfide bond forming amino acids may be either in one or the protein in different
polypeptide chains. Disulfide bonds are formed during post-translational modification of proteins and serve to maintain
protein tertiary and quaternary structures
[8].
This protein has two disulfide bridges connecting the A and B chains and connecting C and D. Due to the fact
that the protein chains are located in similar dimers, we visualized only one located between cysteines chain A and B,
which performs the function of binding and stabilizing these chains.
Hydrogen bonds- a form of association between an electronegative atom and hydrogen atom H, bound
covalently to another electronegative atom. As electronegative atoms can act as N, O or F. Hydrogen bonds may be
intramolecular or intermolecular.
[9].
The hydrogen bond is due to the electrostatic attraction of the hydrogen atom to an electronegative element
having a negative charge. In most cases it is weaker covalent but significantly stronger than usual molecular
attraction to each other in solid and liquid substances. In contrast to the intermolecular hydrogen bond interactions
has properties direction and saturable, so it is often considered a form of a covalent chemical bond. The angle between
the atoms in the fragment (A-B ... H) is usually close to 180°.
[10].
In this protein we studied these hydrogen bonds:
Hydrogen bonds within α-helix.
α-helix - typical secondary structure element of proteins, which has the form of a twisted right helix,
and wherein each amino group (-NH) in the frame forms a hydrogen bond with the carbonyl group (-C = O) amino acid,
which is located an amino acid before 4
[11].
In the applet presented a small part of the protein molecule, which contains α-helix, causing a secondary
structure of a protein.
Hydrogen bonds within ß-sheet.
ß-sheets consist of ß-chains, related sides 2 or 3 hydrogen bonds, forming a slightly twisted, folded
sheets. Higher-level structure formed by a plurality of ß-sheets lead to the formation of protein aggregates
and fibrils observed in many human and animal diseases
[12].
In the applet presented a small part of the protein molecule, which contains & # 223 strands connecting the
two chains A and B together.
Salt bridges, in chemistry, are a combination of two non-covalent interactions: Formation of
hydrogen bonds and electrostatic interactions often observed in proteins, characterized by low entropy values. While
non-covalent interactions are considered to be relatively weak, even such a small contribution to the stabilization of
the protein structure capable of supporting the entire molecule. Salt bridge usually occurs by reaction of the anion
of the carboxyl group of aspartic or glutamic acid and lysine ammine group, or guanidine arginine or histidine. It is
important the distance between the charged residues, it should be not more than 4Å
[13].
This applet represented the salt bridge between the glutamic acid and arginine different circuits that facilitate
the binding of the two chains.
Stacking interaction - a hydrophobic bonds which arise with such an arrangement of aromatic molecules
that resembles a stack of coins in the location and maintained aromatic interactions. Aromatic interactions
(or π-π interaction) - a non-covalent interaction between organic compounds containing aromatic
components. π-π intermolecular interaction caused by overlapping p-orbitals in the π-conjugated
systems, so that they become stronger as the number of π-electrons increases
[14].
We have studied the various aromatic amino acid residues and found stacking interaction in the chain A in two
places between histidine residues. And in one of the stacking interaction of aromatic rings are located precisely above
each other, and in the other - they are symmetrically deployed in space. |
| |
|
LIGAND-BIOMOLECULAR BINDINGS |
|
As previously noted, the composition of alcohol dehydrogenase I comprises 8 of zinc ions, 2 molecules of NAD and
4 molecules of trifluoroethanol. All ligands are distributed uniformly over two similar circuits corresponding dimers.
Thus, all the contacts of the ligand to the protein in a dimer are identical to those in the other dimer. Accordingly,
in our work, we have considered only contacts with the ligands A and B chains of a dimer.
Trifluoroethanol molecule of chain A ([ETF]404:A) is bound to the zinc ion, situated at a
distance 1,78Å of it. Besides zinc ion ([ZN]401:. A) coordinating linked to settling down at a distance 1,95Å
of nitrogen atom of a histidine molecule ([HIS]66:A.NE2), and two sulfur atoms ([CYS]153:A.SG; [CYS]43:A.SG).
The most interesting arrangement of the molecules of trifluoroethanol chain B. It is located next to the zinc ion is
still linked to two sulfur atoms of the cysteines ([CYS]153:V.SG; [CYS]43:V.SG), nitrogen atom of histidine
([HIS]66:B.NE2). However, this chain is coordinating linked to the zinc from oxygen, glutamic acid ([GLU]67:B.OE2), and
an oxygen-trifluoroethanol is not formed. In this case the oxygen atom is further away from the zinc ion, in contrast
to chain A, whereas oxygen from conversely glutamic acid is closer (2,31Å and 3.11Å). Find out what exactly
the connection is held as a part of the protein ligand has not yet succeeded. They were found no covalent bonds,
or hydrogen, or salt bridges were detected.
Another zinc ion is covalently bonded to four sulfur atoms of the cysteines, which are located at a distance
of no more than 2.33Å.
NAD molecules are arranged in chains A and C. According to our assumptions, with the chain ligand connects by
three salt bridges arising between the nitrogen atoms in the molecule NAD and atoms of nitrogen of histidine, lysine
and arginine([8ID]403:A.O2N - [ARG]340:A.NH1; [8ID]403:A.O3B - [LYS]206:A.NZ; [8ID]403:A.O3' - [HIS]48:A.NE2).
Perhaps also a special role in maintaining the structure is played by hydrophobic interactions. As described in the
section on hydrophobic interactions of the molecule NAD is immersed in a large hydrophobic core, the other part
freely visible from the surface.
|
| |
|
|
THE SEARCHING IF CONTACTS
To perform this work, we used the program JMol,
with which we were visualized interactions between protein atoms in the molecule. With the imaging results can be
found on this page. To search for contacts in a protein, we performed the following operations:
For the detection of hydrogen bonds in ß-sheets and α-helix were used commands such as
'select helix' - for α-helix; 'select sheet' - for ß-sheets; 'hbonds on' -
for the direct designation of the hydrogen bonds between the atoms of different amino acid residues.
Disulfide bridge we found by selecting the cysteine residues ([CYS]), between which and perhaps her appearance.
Search hydrophobic cores was carried out with the help of the resource
CluD, which accurately reflects all the core
that are characteristic of that molecule.
To explore surroundings respective low-molecular ligands, we determined the atoms arranged around at
4Å. And then isolated amino acid residues, which are covalently bound ligands, and creating the appropriate applet.
To identify salt bridges formed between the charged amino acid residues, we searched oppositely charged
residues, radicals of which are located at a short distance. Thus two groups of residues were chosen: the positively
charged ([LYS], [HIS], [ARG]) and negatively ([ASP], [GLU]); and each group was marked in a different color, as a
result we were able to identify a specific pair of forming a salt bridge.
Stacking interactions was found by analyzing the mutual arrangement of the various aromatic amino acids
([HIS], [PHE], [TRP], [TYR]) and searching aromatic rings which are arranged parallel at a short distance. |
THE PERSONAL WORK OF TEAM MEMBERS
Page with the report, including: design, presentation of information, editing text, creating a table with
a shared data, creating applets and edit scripts; displaying hydrophobic and ligand-protein interactions in the
molecule, a description of ligand was performed by
Anna Kamysheva.
Creating scripts dedicated to protein-protein interactions, ligands and their interactions with proteins,
the description of protein-protein interactions and contacts search process; the translation work into English
was performed by
Polina Shpudeiko.
|
REFERENCES
- http://www.rcsb.org/pdb/explore.do?structureId=4W6Z
- http://www.uniprot.org/uniprot/P00330
- https://ru.wikipedia.org/wiki/Saccharomyces_cerevisiae
- https://ru.wikipedia.org/wiki/Оксидоредуктазы
- https://ru.wikipedia.org/wiki/Гомотетрамер
- Lehninger Principles of Biochemistry by Albert L. Lehninger, David L. Nelson, Michael M. Cox
- https://ru.wikipedia.org/wiki/Ковалентная_связь
- https://ru.wikipedia.org/wiki/Дисульфидная_связь
- https://ru.wikipedia.org/wiki/Водородная_связь
- http://www.alhimik.ru/stroenie/gl_14.html
- https://ru.wikipedia.org/wiki/Альфа-спираль
- http://hpc.mipt.ru/wp-content/uploads/2012/05/Lecture..
- https://en.wikipedia.org/wiki/Salt_bridge
- https://ru.wikiversity.org/wiki/Стэкинг_взаимодействия
|
| The main page |