Практикум 11. Описание полиморфизмов у пациента
Часть I: подготовка чтений
Для выполнения данного практикума использовались прочтения 8-ой хромосомы пациента. Вначале качество ридов было визулизировано программой FastQC. Затем с конца каждого чтения были удалены нуклеотиды с качеством ниже 20, оставлены только чтения длиной не меньше 50 нуклеотидов. Затем качество ридов было снова визуализировано.
Таблица с командами (исполнялись в папке /nfs/srv/databases/ngs/anton.vlasov/pr11):
| Команда | Что делает |
| fastqc chr8.fastq | Визуализирует качество ридов 8-ой хромосомы. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.fastq chr8_good.fastq TRAILING:20 MINLEN:50 | Удаляет с конца каждого чтения нуклеотиды с качеством ниже 20, оставляет чтения длиной не меньше 50. |
| fastqc chr8_good.fastq | Визуализирует качество улучшенных ридов 8-ой хромосомы. |
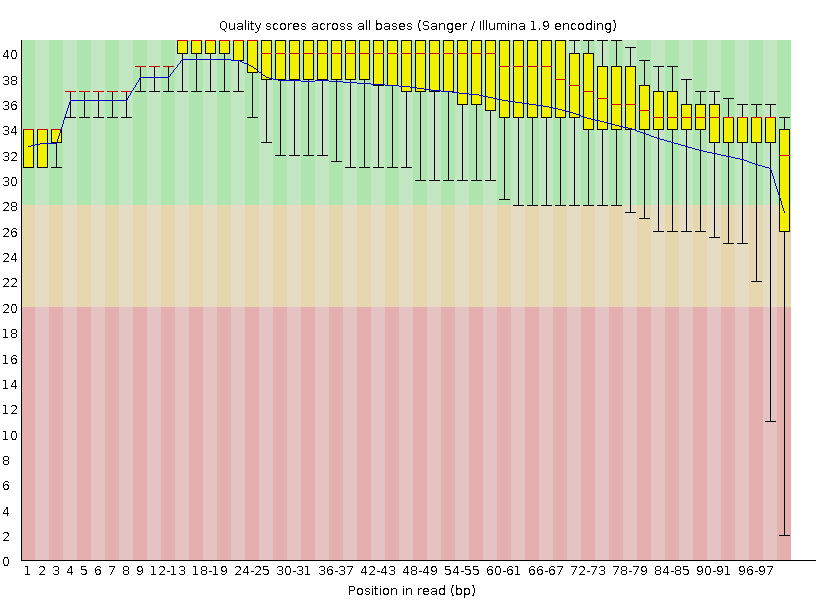
Per base quality до обработки.
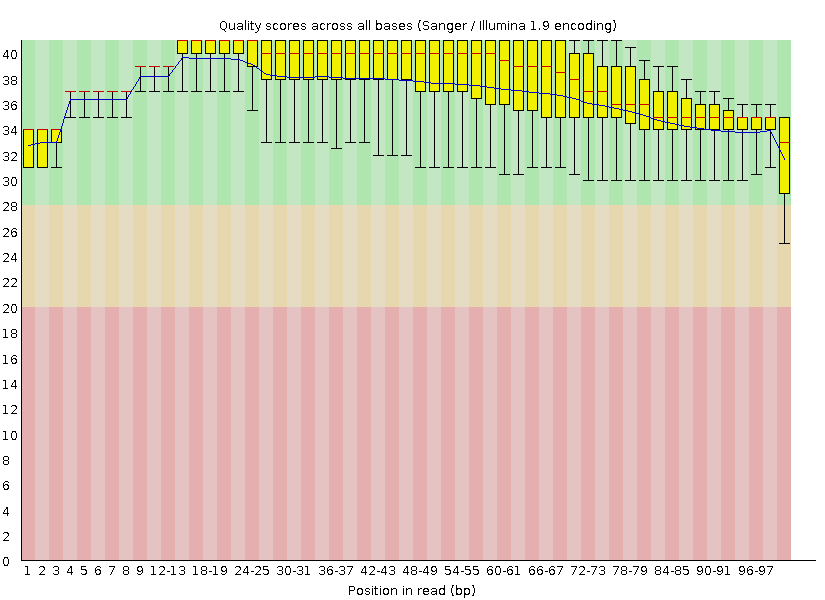
Per base quality после обработки trimmomatic.
Число последовательностей после обработки уменьшилось с 8367 до 8227.
Часть II: картирование чтений
Очищенные чтения были откартированы программой Hisat2. Вначале файл с референсной последовательностью был проиндексирован, а затем было построено выравнивание прочтений и референса в формате .sam. Вывод программы был сохранен в отдельном файле.
Полученный файл с выравниванием был переведен в бинарный .bam формат. Затем выравнивание чтений с референсом было отсортировано по координате в референсе в начале чтения по возрастанию. Затем отсортированный файл был проиндексирован.
Таблица с командами:
| Команда | Что делает |
| hisat2-build chr8.fasta chr8 | Индексирует файл с референсной последовательностью. |
| hisat2 --no-spliced-alignment --no-softclip -x chr8 -U chr8_good.fastq -S align.sam &> result.log | Выравнивает риды с референсной последовательностью, сохраняя информацию о работе программы в файл result.log. |
| samtools view align.sam -b -o align.bam | Преобразует .sam файл в бинарный .bam файл. |
| samtools sort align.bam -T temp.txt -o align_sort.bam | Сортирует по возрастанию выравнивание чтений с референсом по координате в референсе в начале чтения. |
| samtools index align_sort.bam | Индексирует отсортированное выравнивание. |
Из файла result.log можно узнать следующие данные о картировании. 8197 ридов были картированы на референс, 30 ридов не были картированы. Ни один рид не был картирован более одного раза.
Часть III: Анализ SNP
Поиск SNP и инделей
Вначале был получен файл с полиморфизмами в формате .bcf, который затем был преобразован в формат .vcf.
Таблица с командами:
| Команда | Что делает |
| samtools mpileup -uf chr8.fasta align_sort.bam -o SNPs.bcf | Создает файл с полиморфизмами в формате .bcf. |
| bcftools call -cv SNPs.bcf -o SNPs.vcf | Создаёт файл со списком отличий между референсом и чтениями в формате .vcf. |
Примеры полиморфизмов:
| Координаты | Референс | Риды | Тип | Глубина покрытия | Качество |
| 27454785 | TAATGAA | TAA | Делеция | 5 | 58.5 |
| 116631902 | C | A | SNP | 35 | 222.0 |
| 116599199 | T | G | SNP | 45 | 222.0 |
Всего было найдено 5 инделей и 95 SNP.
Аннотация SNP
Файл SNPs.vcf был сконвертирован в формат annovar. Из полученного файла были вручную удалены строки с инделями. Затем полиморфизмы были проаннотированы по различным базам данных.
Таблица с командами:
| Команда | Что делает |
| convert2annovar.pl --format vcf --outfile SNPs.annovar SNPs.vcf | Конвертирует файл SNPs.vcf в формат annovar. |
| annotate_variation.pl -filter -out annots/snp138 -build hg19 -dbtype snp138 SNPs.annovar /nfs/srv/databases/annovar/humandb.old/ | Разделяет полиморфизмы по наличию в dbsnp. |
| annotate_variation.pl -out annots/refgen -build hg19 SNPs.annovar /nfs/srv/databases/annovar/humandb.old/ | Определяет положение полиморфизма в геноме, а также функцию полиморфзма при её наличии. |
| annotate_variation.pl -filter -out annots/1000genomes -build hg19 -dbtype 1000g2014oct_all SNPs.annovar /nfs/srv/databases/annovar/humandb.old/ | Определяет частоты полифморфизмов по базе данных 1000 genomes. |
| annotate_variation.pl -regionanno -out annots/gwas -build hg19 -dbtype gwasCatalog SNPs.annovar /nfs/srv/databases/annovar/humandb.old/ | Определяет признаки, которые кодируют данные полиморфизмы по базе данных GWAS. |
| annotate_variation.pl -filter -out annots/clinvar -build hg19 -dbtype clinvar_20150629 SNPs.annovar /nfs/srv/databases/annovar/humandb.old/ | Аннотирует полиморфизмы по базе данных Clinvar |
Результаты:
- Качество и число чтений полиморфизмов:
- Среднее качество: 66,7; число чтений: 14,5.
- Медиана качества: 11,3; числа чтений: 2.
- Верхний квартиль качества: 125; числа чтений: 16.
- Всего 42 полиморфизма с качеством чтений больше 25, 35 полиморфизмов были прочитаны более 2 раз, 44 полиморфизма были прочитаны только 1 раз (качество чтений таких полиморфизмов не превышает 11,3).
- Из 95 полиморфизмов 77 имеют rs.
- RefGene делит полиморфизмы на следующие категории (в скобках число таких полиморфизмову пациента):
- UTR3 (13) - 3'-нетранслируемая область мРНК
- intronic (60)
- exonic (5)
- intergenic (17) - межгенные участки ДНК
- Полиморфизмы обнаружены в экзонах следующих генов (в скобках замена нуклеотида: замена аминокислоты):
- CLU → белок кластерин (clusterin). Синонимическая замена (T789 → C: H263 → H).
- HNF4G → ядерный фактор гепатоцитов 4-гамма (hepatocyte nuclear factor 4 gamma). 2 несинонимические и 1 синонимическая замены (G86 → A: S29 → N; G627 → A: L209 → L; G681 → A: M227 → I).
- TRPS1 → фактор трихо-рино-фалангеального синдрома I типа (tricho-rhino-phalangeal syndrome Type 1 factor). 1 синонимическая замена (G423 → T: P141 → P или G402 → T: P134 → P или G396 → T: P132 → P).
- Ни один из полиморфизмов не был найден в базе данных Clinvar, поэтому про клиническую аннотацию сделать выводы нельзя.
- По данным GWAS некоторые полиморфизмы повышают вероятность возникновения болезни Альцгеймера, один полимофизм отвечает за содержане солей мочевой кислоты, ещё один - за содержание холестерола высокой плотности (HDL cholesterol).
- Аннотирование по базе данных 1000Genomes:
- 21 полиморфим не был найден в базе данных. У 18 из них качество чтений не превышает 11,3.
- Средняя встречаемость найденных полиморфизмов: 46,33%
- Медиана: 53,82%.
- Верхний квартиль: 67,44%.
- Минимум: 0,5%.
- Максимум: 100% (хотя не очень понятно, почему тогда это является полиморфизмом).
- По результатам аннотирования была составлена сводная таблица. Скачать таблицу.