Ресеквенирование. Поиск полиморфизмов у человека
1. Подготовка чтений
Для хромосомы №20 были скопированы следующие файлы:
chr20.fasta
chr20.fastq
И с помощью FastQC проанализировано качество чтений:
fastqc chr20.fastq
Отчёт программы:
chr20_fastqc_before.zip
Картинка из отчёта:
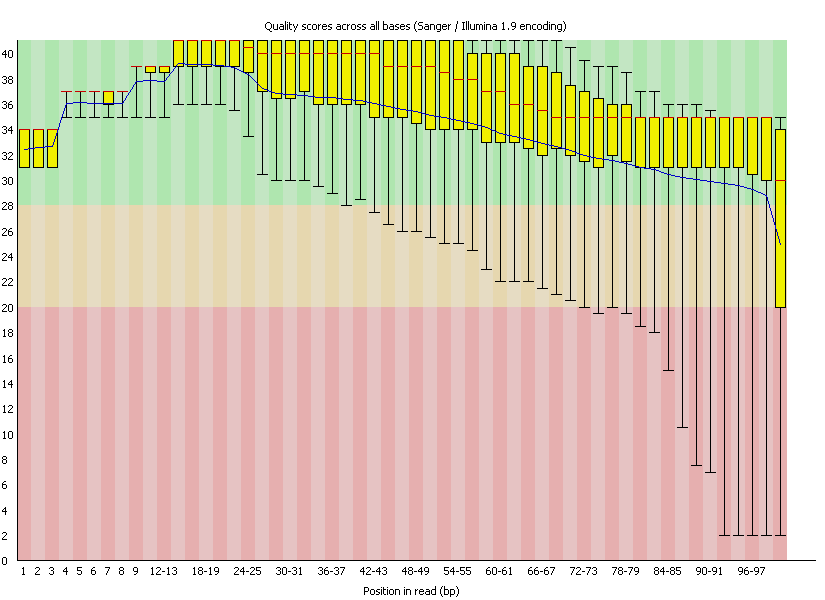
Далее с помощью программы trimmomatic произведена коррекция исходного качества:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr20.fastq chr20_trimmed.fastq
TRAILING:20 MINLEN:50
TrimmomaticSE: Started with arguments: -phred33 chr20.fastq chr20_trimmed.fastq
TRAILING:20 MINLEN:50
Automatically using 8 threads
Input Reads: 4661 Surviving: 4472 (95,95%) Dropped: 189 (4,05%)
TrimmomaticSE: Completed successfully
arma@kodomo:~/term3/block3/pr13$
По выводу видим, что удалилось 189 чтений длиной менее 50 нуклеотидов, включая те, что получились после удаления нуклеотидов с качеством ниже 20 с конца чтения. Осталось 4472 чтения.
Результат коррекции:chr20_trimmed.fastq
Анализ качества чтений после коррекции исходного файла:
Файл с отчётом:chr20_trimmed_fastqc.zip
Картинка из отчёта:
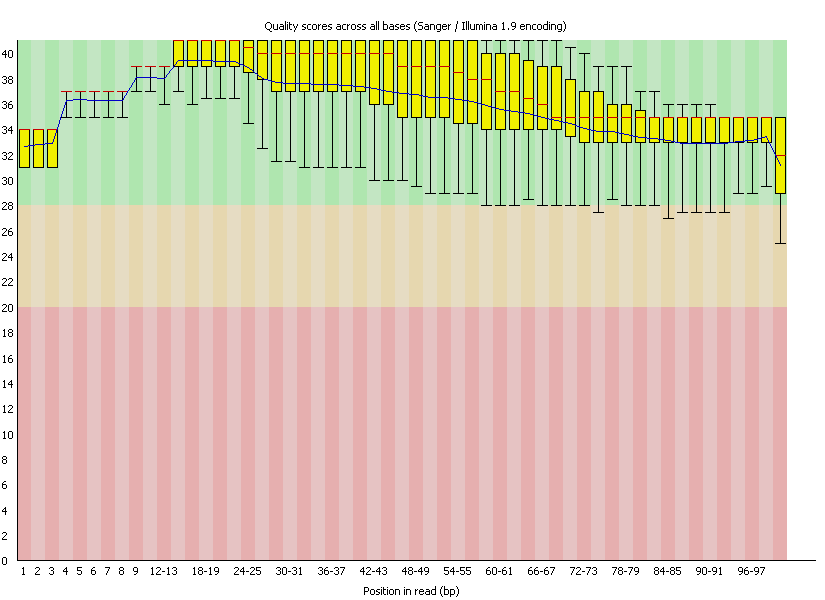
2. Картирование чтений
Индексация файла: bwa index chr20.fasta
Выравнивание индексированного файла и откорректированных прочтений: bwa mem chr20.fasta chr20_trimmed.fastq > chr20_bwa.sam
Анализ выравнивания:
samtools view -b chr20_bwa.sam -o sam_outf.bam # получаем нужный формат
samtools sort -o sam_sort.bam -T out.prefix sam_outf.bam #сортировка по координате в референсе
samtools index sam_sort.bam # индексация сортированного файла
samtools idxstats sam_sort.bam # статистика картирования чтений
Скриншот статистики:

Из 4472 чтений откартировалось 4468, 4 не нашли совпадений с картой генома.
Анализ однонуклеотидных полиморфизмов (SNP)
samtools mpileup -uf chr20.fasta sam_sort.bam > snp.bcf # поиск полиморфизмов
bcftools call -cv snp.bcf > dif.vcf # вывод списка отличий в файл
Чтобы визуально оценить список, смотрим описание формата vcf, прилагающееся к руководству bcftools (http://samtools.github.io/hts-specs/) и видим, что начиная со строки с одой решёткой в начале его можно сгрузить в Excel. Что и было сделано.
vcf-файл отличий
excel-файл отличий
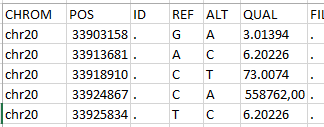
Инделей не обнаружено (в графе "INFO" только "DP")
Аннотация по 5 базам данных:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 dif.vcf > dif.avinput
perl /nfs/srv/databases/annovar/annotate_variation.pl -out RefGeneAnnotation -build hg19 dif.avinput /nfs/srv/databases/annovar/humandb/
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out SNP138Annotate -build hg19 -dbtype snp138 dif.avinput /nfs/srv/databases/annovar/humandb/
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_eur -buildver hg19 -out 1000gAnnotate dif.avinput /nfs/srv/databases/annovar/humandb/
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out gwasAnnotate -dbtype gwasCatalog dif.avinput /nfs/srv/databases/annovar/humandb/
В итоге каждого скрипта получим 2-3 файла: .log его работы и найденные (filtered)/не найденные(dropped) в базе записи.
Составим по ним сводную таблицу аннотации: excel-файл
В клинической базе данных gwas аннотированы 3 snp, связанные со следующими паталогиями:
34025756: A на G, аномалия роста
48522330: G на A, развитие псориаза
56190634: С на T, атрофия гиппокампа
RefSeq делит замены по участкам расположения(экзонные, интронные) и в экзонных (у меня получилось их 8) выделяет, синонимична ли замена.
rs есть у 30 snp; частоту встречаемости можно оценить по результатам выдачи 1000g (см. таблицу)
|