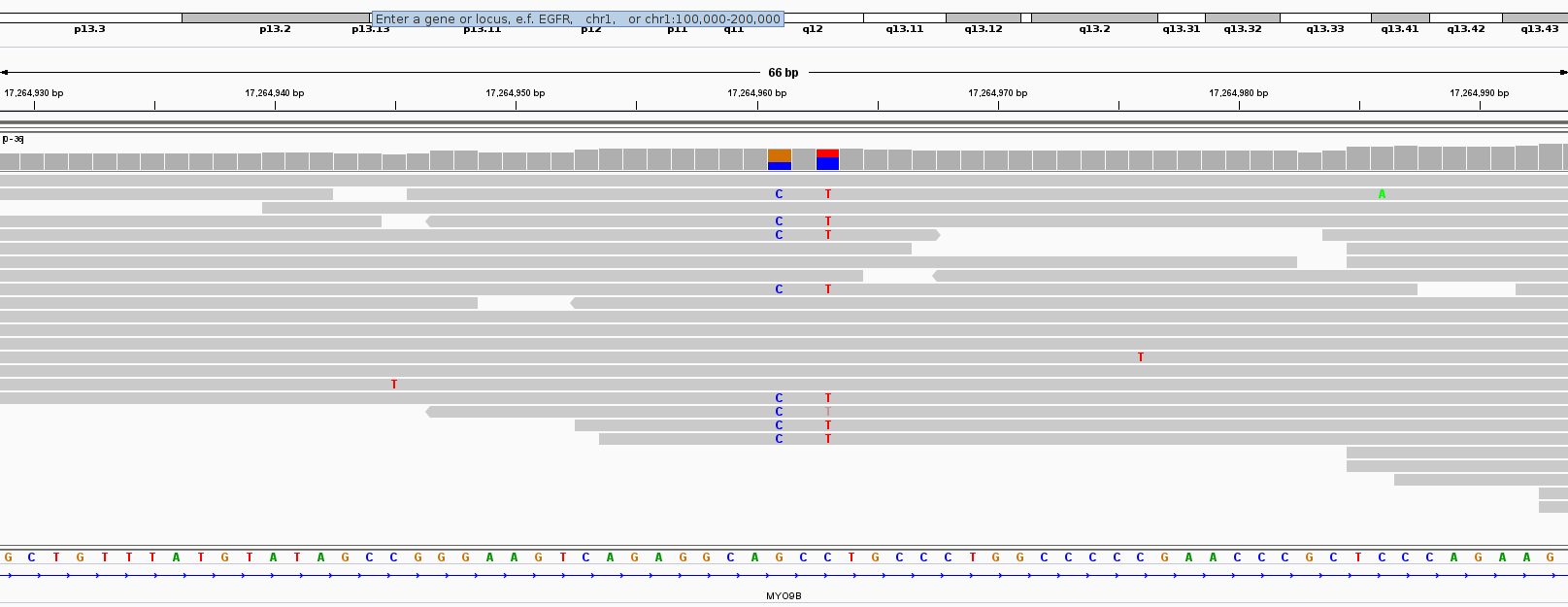
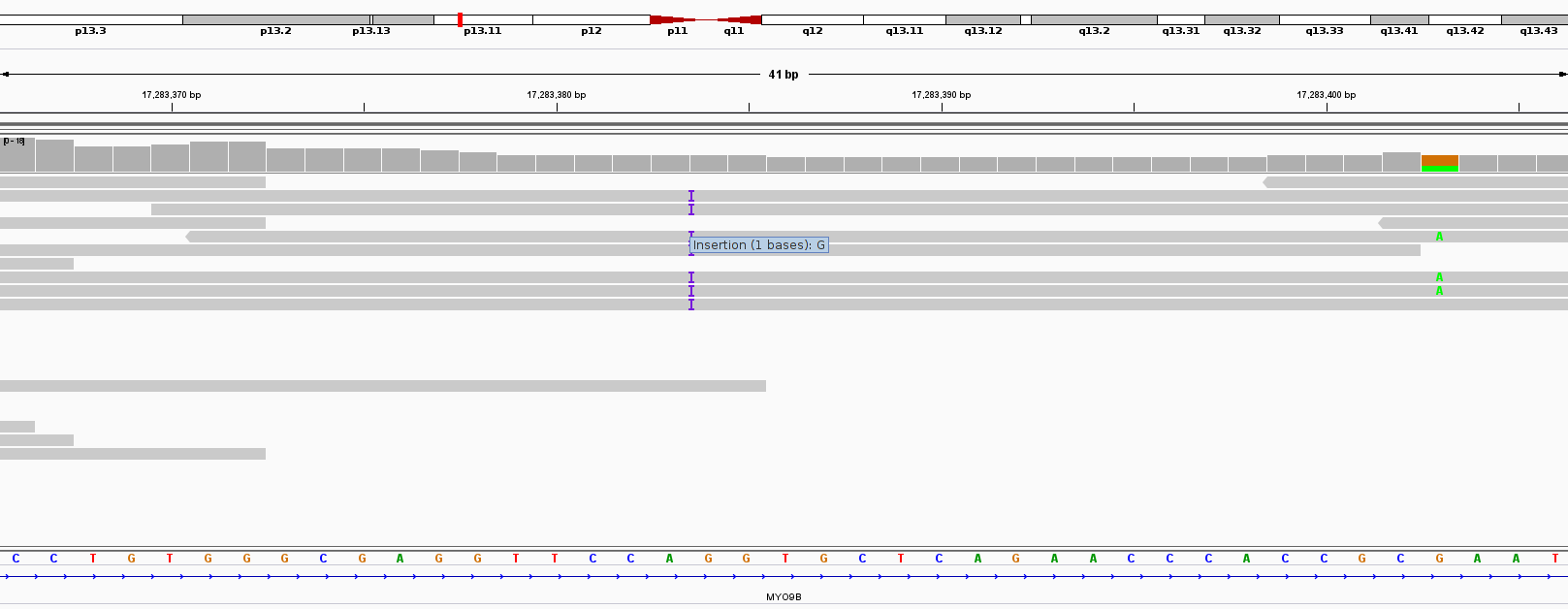
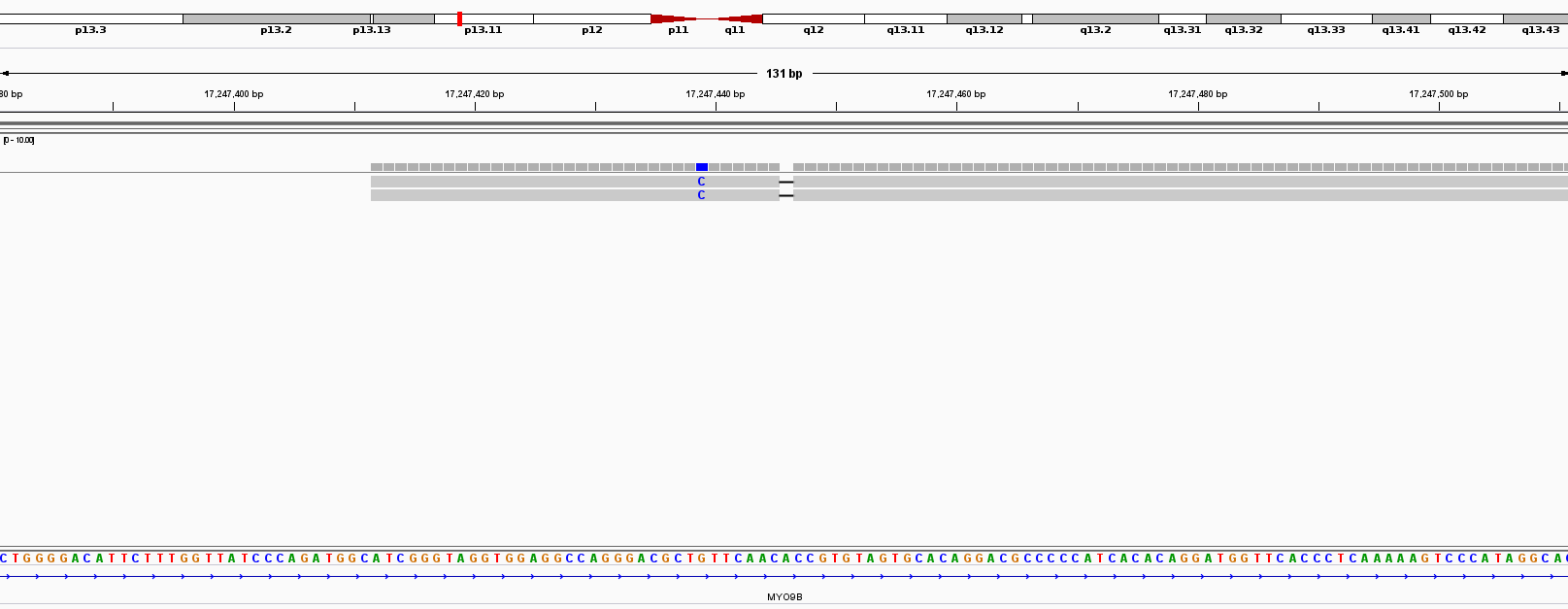
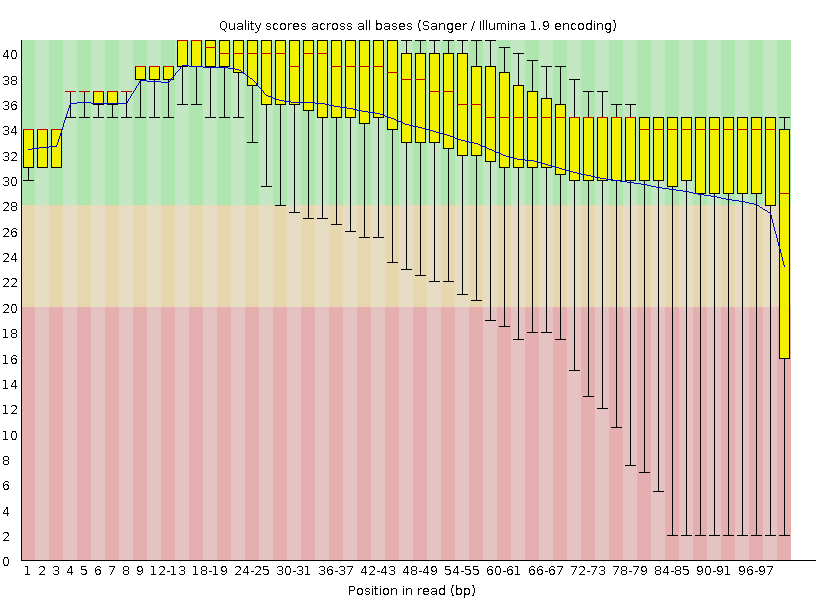
 Рисунок 1. Выдача программы FastQC с неочищенными ридами
Рисунок 1. Выдача программы FastQC с неочищенными ридами
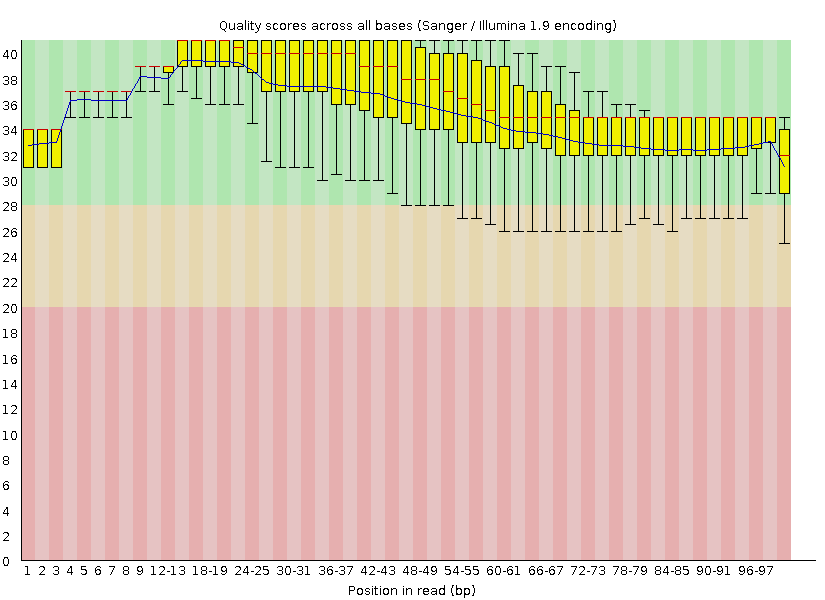 Рисунок 2. Выдача программы FastQC с очищенными ридами
Рисунок 2. Выдача программы FastQC с очищенными ридами
Число чтений до чистки: 5524;
Число чтений после чистки: 5227;
Отсеялись все чтения длиной качественной части менее 50.
Остальная выдача FastQC имеет незначительные различия; упоминания стоит лишь распределение последовательностей по длине:
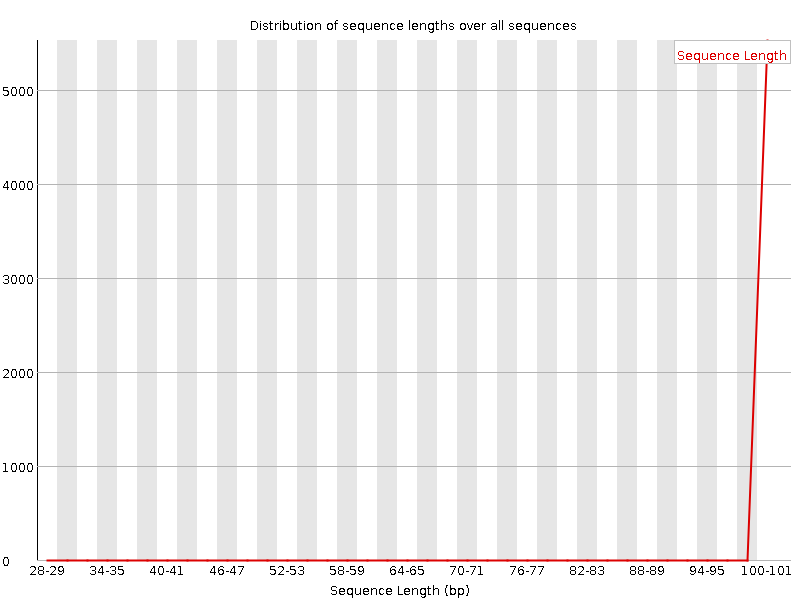 Рисунок 3. Распределение длин неочищенных ридов
Рисунок 3. Распределение длин неочищенных ридов
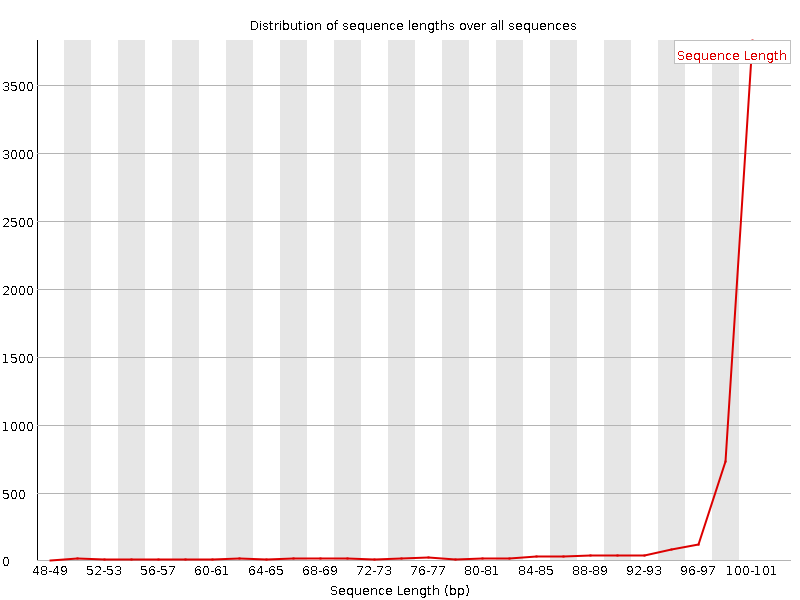 Рисунок 4. Распределение длин очищенных ридов
Рисунок 4. Распределение длин очищенных ридов
Из этой выдачи видно, что последовательности длиной более 100 часто укорачивались из-за низкокачественных концевых участков, а последовательности длиной менее 50 нуклеотидов выбрасывались.