| Была создана директория /nfs/srv/databases/ngs/asya.kalashnikova, куда были скопированы файлы chr8.fastq и chr8.fasta - с ридами и хромосомой. Далее производился анализ качества чтений с помощью программы FastQC на Kodomo. Была использована команда (1). В итоге были получены 2 файла: chr8_fastqc.html и chr8_fastqc.zip . Проводилась очистка чтений с помощью программы Trimmomatic, причем необходимо оставить только чтения длиной не меньше 50 и отрезать с конца каждого чтения нуклеотиды с качеством ниже 20. |
|
Использовались определенные операции:
1) TRAILING: отрезает основания в конце прочтения, качество которых будет ниже заданного значения; 2) MINLEN: убирает чтения, чья длина менее заданной. |
|
(1) fastqc chr8.fastq
(2) java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr8.fastq 1.fastq TRAILING:20 MINLEN:50 (3) fastqc 1.fastq |
|---|
|
Далее был проведен анализ качества очищенных чтений (файл: 1.fastq) с помощью команды (3).
В итоге было получено 2 файла: 1_fastqc.html и 1_fastqc.zip .
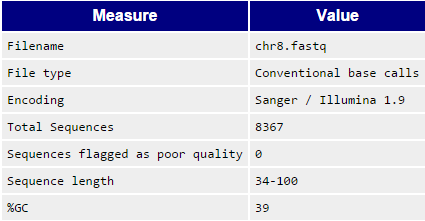
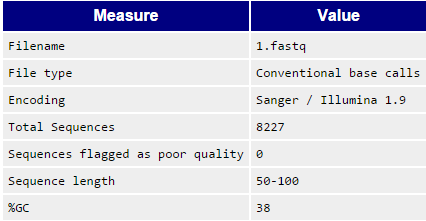
Число чтений до и после чистки: 8367 (до), 8227 (после). |
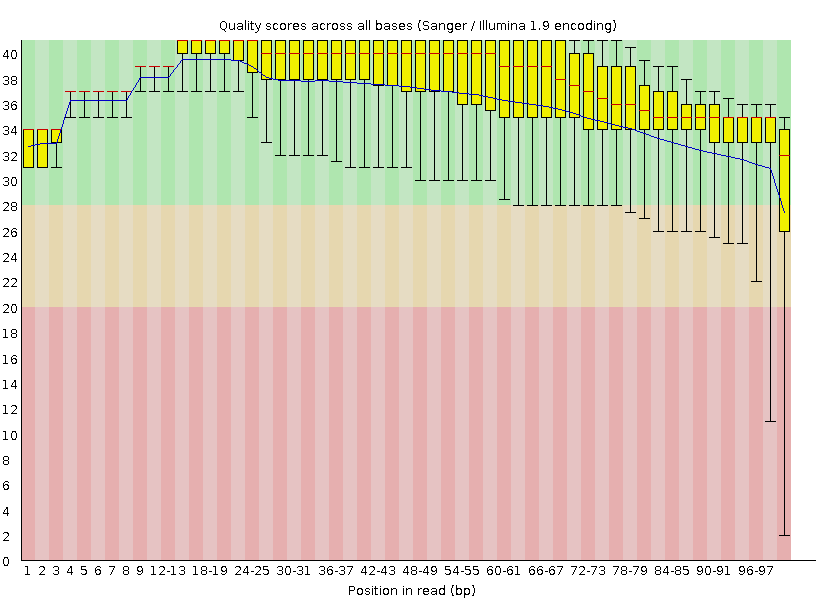
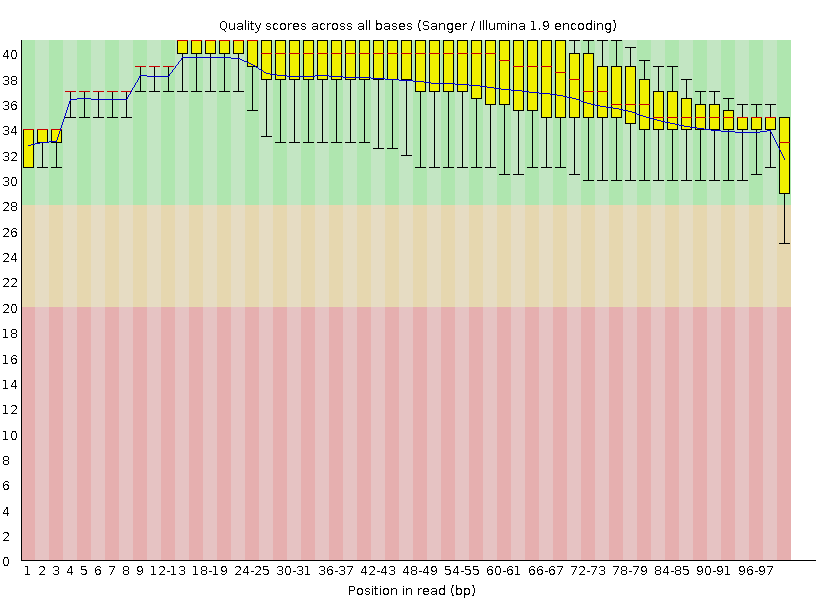
Рис. 1 - Per base sequence quality (до и после чистки) - увеличение при нажатии
| |
|
Рис. 2 - Basic Statistics (до и после чистки) - увеличение при нажатии
 |
 |
|
Как можно увидеть на рис.1 очистка существенно улучшила показатель качества ридов, к тому же подняла ценз на длину - как и предполагалось (см. рис. 2). Показатель "GC Content" практически не изменяется. Показатель длины чтений до чистки составлял 34-100, после очистки - 50-100. Таким образом, отсев ридов происходил, как и предполагалось по качеству и длине рида. |
|
1) Необходимо было откартировать чтения с помощью программы BWA.
Для этого необходимо было проиндексировать референсную последовательность с помощью команды (1).
Затем было построено выравнивание прочтений и референса в формате sam с помощью алгоритма mem команды (2).
2) Далее происходил анализ выравнивания: сначала выравнивание было переведено в бинарный формат bam с помощью пакета samtools, команды (3). Опция -o была использована для указания выходного файла, -b - для смены формата с sam на bam. Получившийся файл был отсортирован по координате в референсе начала чтения - команда (4). Далее файл sort.bam был проиндексирован командой (5) и исследован на предмет числа откартированных чтений на геном с помощью команды (6). Был получен файл chr8_car.txt . Число не картированных/картированных чтений на хромосому: 3/8224 из 8227. |
|
(1) bwa index chr8.fasta
(2) bwa mem chr8.fasta 1.fastq > align.sam (3) samtools view align.sam -b -o align.bam (4) samtools sort align.bam sort (5) samtools index sort.bam (6) samtools idxstats sort.bam > chr8_car.txt |
|---|
| 1) Поиск SNP и инделей: с помощью команды (1) был создан файл с полиморфизмами с формате bcf, а с помощью (2) был создан файл со списком отличий между референсом и чтениями в формате vcf. Был получен файл read.vcf. Число snp: 94; число инделей: 8. Далее необходимо было найти и описать 3 полиморфизма из vcf файла - таблица 1. |
Таблица 1. Описание полиморфизмов
| Полиморфизм | Координата | Качество чтений | Глубина покрытия | Было в референсе | Найдено в чтениях | Тип полиморфизма |
| №1 | 27466315 | 154.998 | 29 | T | C | Замена |
| №2 | 116424270 | 59.456 | 31 | A | AC | Вставка |
| №3 | 76452313 | 221.999 | 30 | G | A | Замена |
|
Покрытие: min - 1; max - 134; среднее - 14,8.
Качество: min - 3,01618; max - 225,009; среднее - 68,1076. (Я не совсем представляю себе критерии "хороших" покрытия и качества, но предполагаю, что среднее значение качества являет довольно неплохим, в то время как среднее значение глубины покрытия я бы назвала "средним"). SNP привели к данным нуклеотидным заменам: T - (C, A, G); C - (T, G, A); G - (A, T); A - (G, T, C). 2) Аннотация SNP: с помощью annovar необходимо было проаннотировать полученные snp по базам данных refgene, dbsnp, 1000 genomes, GWAS, Clinvar. Сначала был получен файл без инделей: No_indels.vcf. Потом было необходимо получить файл формата, с которым умеет работать annovar с помощью скрипта convert2annovar.pl, который был скопирован в директорию с остальными файлами. Это было сделано командой (3). Dbsnp: аннотация была получена с помощью команды (4). Данная база используется для того, чтобы узнать, какие из snp имеют rs. Аннотация по этой базе данных основана на фильтрации (filter-based annotation). На выходе были получены 3 файла: dbsnp.hg19_snp138_dropped (файл с snp, имеющими rs), dbsnp.hg19_snp138_filtered (файл с snp, не имеющих rs), dbsnp.log (файл, содержащий информацию по запуску). Таким образом, 76 snp из 94 имеют rs (18 не имеют). Refgene: аннотация была получена с помощью команды (5). Аннотация по этой базе данных основана на генной разметке (gene-based annotation). На выходе 3 файла: refgene.exonic (файл содержит snp, найденные в экзонах), refgene.log (файл с информацией о запуске и работе программы), refgene.variant_function (файл, содержащий все snp). Категории: 1) Exonic: 5; 2) Intronic: 58; 3) Intergenic: 18; 4) UTR3: 13. Гены, в которые попали snp: TRPS1 (exonic, UTR3, intronic), CLU (UTR3, intronic, exonic), CASC9 (intergenic), HNF4G (intergenic, intronic, exonic, UTR3). 1000 genomes: аннотация была получена с помощью команды (6). Аннотация по этой базе данных основана на фильтрации (filter-based annotation). Выход похож на выход dbsnp, но по базе 1000 genomes. На выходе получены 3 файла: 1000gen.hg19_ALL.sites.2014_10_filtered, 1000gen.log (файл с информацией по запуску), 1000gen.hg19_ALL.sites.2014_10_dropped. Аналогично, не было аннотировано 18 snp. Частота snp: максимальная - 1; минимальная - 0,00499201; средняя - 0,466446. Gwas: аннотация была получена с помощью команды (7). Аннотация по этой базе данных основана на разметке других регионов генома (region-based annotation). Данная база аннотирует snp, ассоциируемые с определенными заболеваниями, то есть база отвечает за клиническую аннотацию. На выходе было получено 2 файла: gwas.log (файл с информацией по запуску), gwas.hg19_gwasCatalog (файл с информацией по болезням). Было аннотировано 4 snp: 2 ассоциируемы с болезнью Альцгеймера, 1 - с "хорошим" холестерином (HDL cholesterol), 1 - с уровнем мочевой кислоты. Clinvar: аннотация была получена с помощью команды (8). Аннотация по этой базе данных основана на фильтрации (filter-based annotation). На выход получаем 3 файла, аналогичные выходу dbsnp, но в данном случае аннотированных snp не наблюдается. Файл с параметрами запуска: clinvar.log; файл с неанотированными snp (всеми): clinvar_filtered. Таблица snp с аннотациями по базам данных |
|
(1) samtools mpileup -uf chr8.fasta sort.bam -o polymorf.bcf
(2) bcftools call -cv polymorf.bcf -o read.vcf (3) perl convert2annovar.pl -format vcf4 No_indels.vcf -outfile No_indels.avinput (4) perl annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 No_indels.avinput /nfs/srv/databases/annovar/humandb (5) perl annotate_variation.pl -out refgene -build hg19 No_indels.avinput /nfs/srv/databases/annovar/humandb (6) perl annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000gen No_indels.avinput /nfs/srv/databases/annovar/humandb (7) perl annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog No_indels.avinput /nfs/srv/databases/annovar/humandb (8) perl annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar No_indels.avinput /nfs/srv/databases/annovar/humandb |
|---|
© Kalashnikova Anastasia, 2016